伴发作性中枢神经系统障碍腓骨肌萎缩症X1型1例及文献复习
2023-07-08戎丹燕刘卫国郭志颖
戎丹燕 刘卫国 于 淼 郭志颖 周 昊
南京医科大学附属脑科医院,江苏 南京 210029
腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)的遗传方式可分为常染色体显性遗传、常染色体隐性遗传以及X 连锁遗传[1-2]。在CMT 中,X 连锁的腓骨肌萎缩症X1型(Charcot-Marie-Tooth type 1,CMTX1)是第二常见的类型,占所有CMT 患者的7%~15%[3]。CMTX1 是由于染色体Xq13.1 上的缝隙连接β1(gap junction protein beta 1,GJB1)基因突变所致,为X连锁显性遗传,男性症状明显,女性也可发病,但症状较轻,大多数患者在7~26 岁发病[4]。大部分CMTX1患者与其他CMT类似,临床表现为典型的周围神经受累症状,如进行性远端为主肌肉萎缩、无力和感觉异常,腱反射减弱或消失,CMTX1患者周围神经的神经电生理和病理学检查多表现为脱髓鞘改变伴明显轴索变性[5]。CMTX1的特殊性在于发作性中枢神经系统功能障碍及脑白质病变。本文结合1 例CMTX1病例重点分析该类患者发作性中枢神经系统症状的临床特征及遗传学特点,以期提高神经内科医生对本病发作性症状的认识。
1 病例资料
患者男,18 岁,高中在读学生,因“发作性肢体麻木无力,言语不清”于2021-08-09 入院。患者2021-08-08 夜间突然出现右侧上下肢发麻感,症状持续时间约10 余分钟后好转,当晚再次出现上述症状,伴言语不清,左侧上下肢轻度发麻和无力,症状持续存在。至急诊,当时测体温38 ℃,后随着体温恢复正常患者伴随的肢体麻木无力、言语不能等症状亦自行好转。患者本次发病前数天有劳累、腹泻史。
既往史及家族史:患者于2015年(14 岁)因双下肢行走不稳就诊于南京市儿童医院,临床考虑CMT,基因检测发现CMTX1 致病基因为GJB1c.65G>A(p.Arg22Gln),其母亲为杂合子变异,仅表现弓形足,外婆亦有弓形足,外婆的弟弟先天性跛足,家系图见图1,考虑诊断为CMTX1。患者曾在2019 年因肢体麻木、言语不清就诊本院。头颅MRI(图2A)提示胼胝体压部、双侧半卵圆中心及放射冠区多发异常信号。当时诊断为“急性播散性脑脊髓炎”,给予激素冲击治疗,患者第2 天肢体麻木、言语不清等症状完全缓解,4 个月后复查头颅MRI(图2B)提示脑内病灶完全消失。患者此次发病1 个月前于外院行马蹄足矫形术。

图1 先证者家系图Figure 1 Family pedigree of the proband
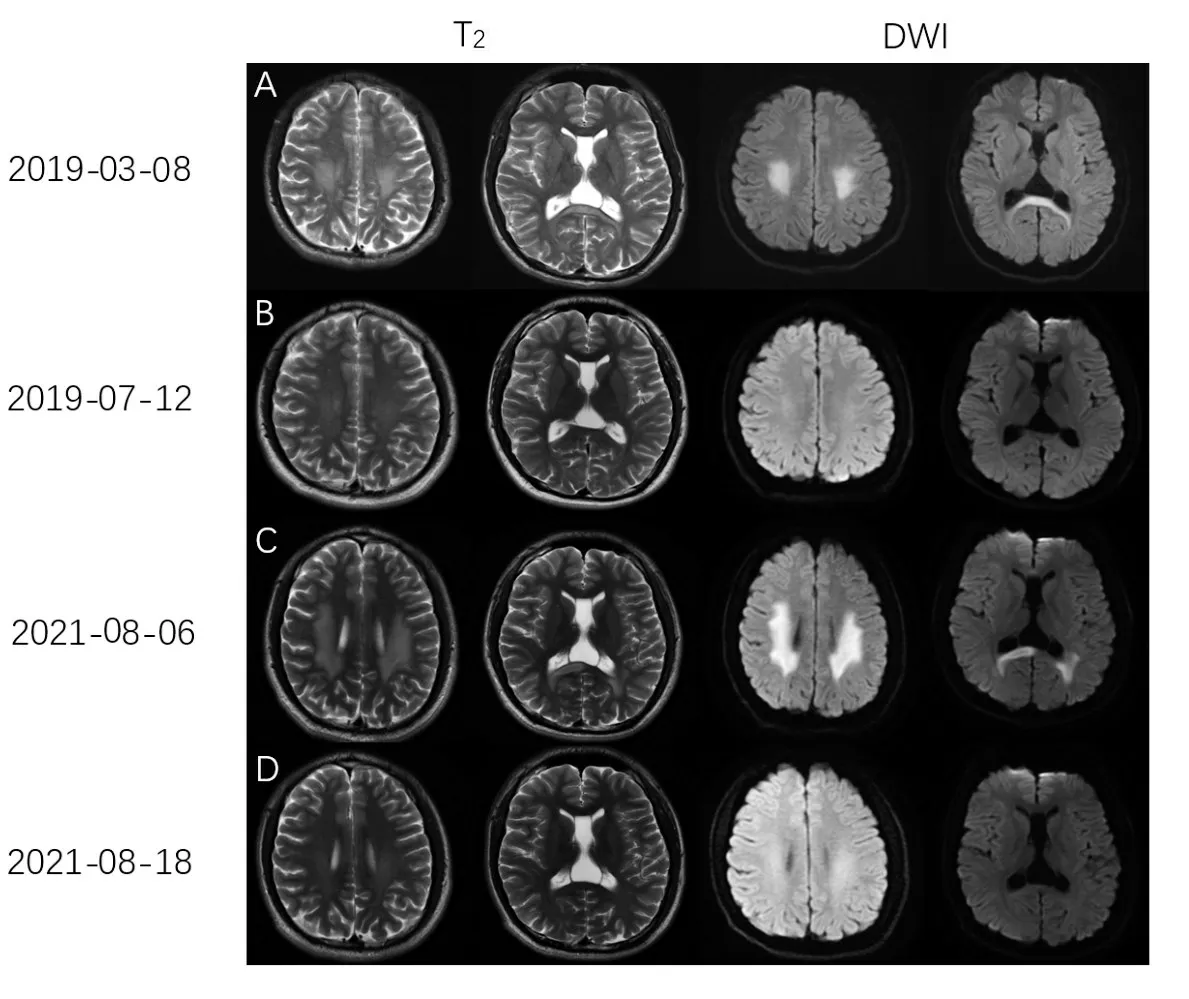
图2 A、B为第1次发作期影像,C、D为第2次发作期影像,A、B、C、D分别显示患者两次发病急性期及缓解期头颅MRI。左边两列是T2加权像,右边两列是DWI加权像。A、C示先证者急性发作期,DWI显示对称性脑室旁白质高信号,累及胼胝体压部,且第2次范围较大。B、D示先证者发病恢复期头颅MRI(T2像)复查,脑白质异常信号明显消退Figure 2 Magnetic resonance imaging(MRI)of the brain, A, B, C, and D showed the patient’s two brain MRIs in the acute phase and remission phase, respectively.The left two columns are T2-weighted sequences and the right two columns are diffusion-weighted sequences.A and C showed the acute exacerbation of the proband, DWI showed symmetrical periventricular white matter hyperintensity, involving the corpus callosum, and the second episode showed larger lesion.B and D showed the re-examination of brain MRI in the convalescent period of the proband, and the abnormal signal of the white matter has obviously subsided
入院体格检查(患者处于中枢神经系统症状发作期):神清,精神差,右侧鼓腮无力,左侧鼻唇沟偏浅,言语欠清,说话费力,伸舌左偏,双手大鱼际肌不饱满,双下肢膝关节以下肌肉略有萎缩。四肢肌力Ⅴ-,四肢肌张力偏低,四肢腱反射(±),右侧巴宾斯基征(-),左侧巴宾斯基征无法查(安装弓形足矫形器),右侧指鼻试验欠稳准,左侧指鼻试验基本稳准,双侧跟膝胫试验无法查,右侧上下肢针刺觉减退,颈软无抵抗,屈颈时自诉有不适感,凯尔尼格征(-)。患者发作性症状消失后除肌肉萎缩、四肢腱反射(±)、弓形足(图3)等体征外,余阳性体征均恢复正常。

图3 先证者右侧足部照片Figure 3 Picture of the proband’s right foot
诊断与治疗经过:入院第1 天给予丙种球蛋白25 g 静滴,第2 天中午出现呼吸困难、饮水呛咳等症状,紧急联系外院行支架矫形器拆除术,术后急查头颅MRI(图2C)示双侧半卵圆中心、放射冠、胼胝体压部见多个点状稍长T1、稍长T2异常信号,DWI序列高信号。临床诊断疑似CMTX1 伴发作性中枢神经系统症状和脑白质病变,立即停用丙种球蛋白,给予营养神经、吸氧、心电监护等对症治疗。当晚患者呼吸困难、饮水呛咳症状好转,后住院10 d内未再出现发作性症状,复查头颅MRI(图2D)示颅内异常信号基本消失。患者出院半年内两次随访均未出现发作性症状,均未再复发。
其他辅助检查:神经肌电图检查(2021-08-06,表1)提示四肢周围神经存在脱髓鞘及轴索损害。诱发电位见:(1)双侧正中神经至皮层感觉传导通路功能障碍(皮层间);(2)双侧胫后神经至皮层感觉传导通路功能障碍;(3)双侧脑干听觉传导通路功能障碍;(4)双侧视觉传导通路功能障碍。脑电图(2021-08-06,图4B)示广泛中度传导异常(双侧枕、顶及后颞区偶见尖慢波单发一次,双侧大致同步对称)。脑脊液(2019-03-11):(1)脑脊液生化:蛋白0.43 g/L,IgG 43.8 mg/L,白蛋白226 mg/L;(2)脑脊液细胞学:白细胞1 个/μL,淋巴细胞70%,单核细胞30%;(3)脑脊液病理细胞学检测未见明显异常。其余血尿便常规、生化、肝肾功能、凝血检测、甲状腺功能、自身抗体、维生素组合、红细胞沉降率、中枢神经系统脱髓鞘相关抗体6 项检测(本次血标本)均未见明显异常。

表1 腓骨肌萎缩症X1型1例患者两次神经肌电图检查结果Table 1 Results of two electromyographic examinations of a patient with Charcot-Marie-Tooth disease type X1

图4 A:第1次发作期蝶骨电极脑电图;B:第2次发作期蝶骨电极脑电图。第2次发作较第1次出现双侧枕、顶及后颞区偶见尖慢波单发1次,双侧大致同步对称Figure 4 A shows the electroencephalography(EEG)of the sphenoid bone electrode during the first dysfunction, and B shows the EEG of the sphenoid bone electrode during the second dysfunction.The second episode is occasionally sharp slow wave in the bilateral occipital, parietal, and posterior temporal regions, and the bilateral sides are roughly synchronous and symmetrical
2 讨论
CMTX1 是涉及周围神经系统病变的遗传性疾病。近年来,越来越多的文献报道了该病发作性中枢神经系统障碍的特点,如短暂性卒中样症状及可逆性脑白质病变,考虑到该表型并不常见,本文结合2021年脑科医院收集的1例伴发作性中枢神经系统障碍的CMTX1患者的临床资料及遗传学特点,结合TIAN 等[6]总结的47 例病例的英文综述及中文报道的10例CMTX1患者信息(表2)加以总结分析。
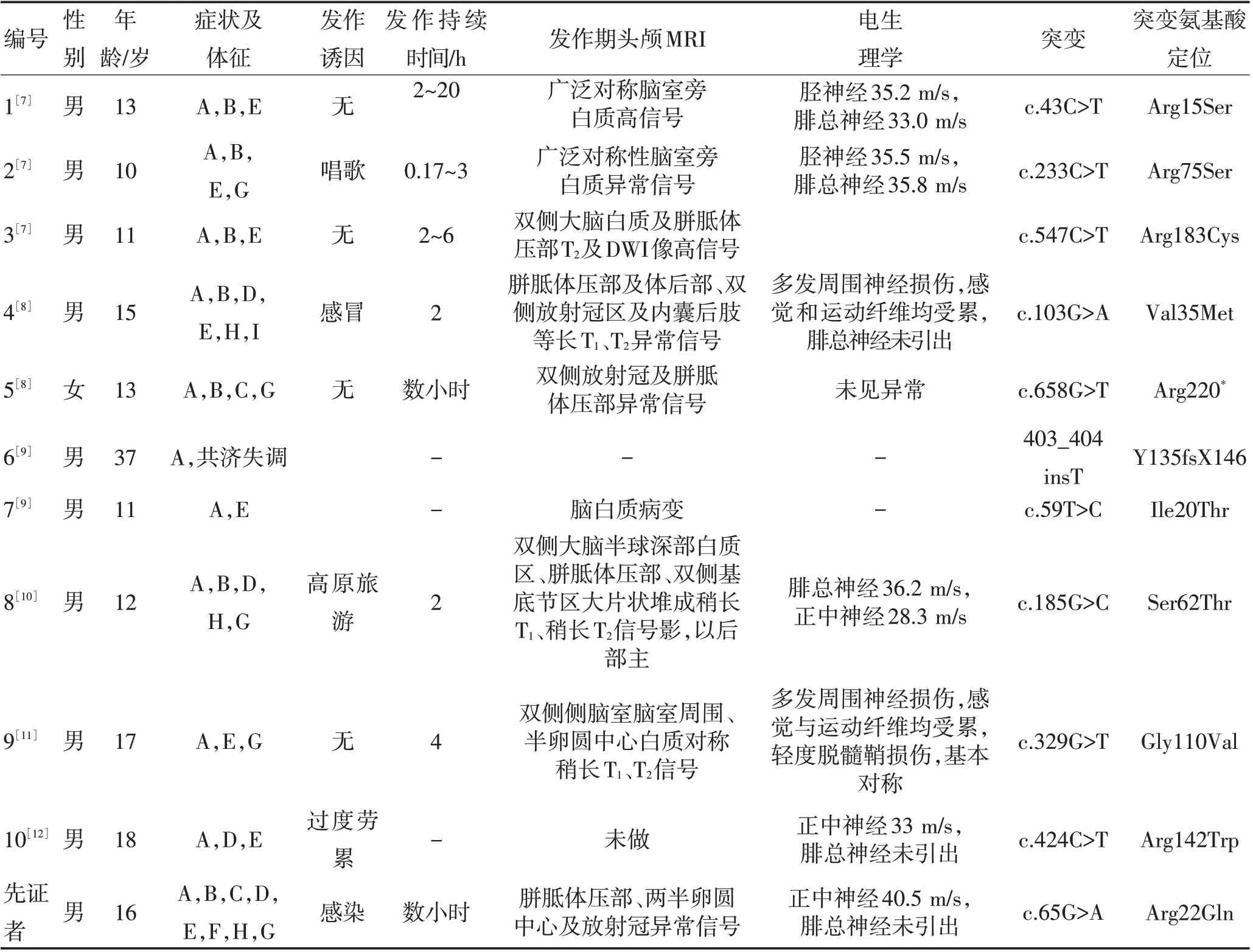
表2 发作性中枢神经系统功能障碍的CMTX1患者的临床表现和基因型Table 2 Clinical manifestations and genotypes of CMTX1 patients with episodic central nervous system dysfunction
58例病例中,3例女性,55例男性。首次发作的平均年龄12.2 岁。诱因:前3 位是感染或发热15 例(25.9%),高原旅游史7 例(12.1%),剧烈运动4 例(6.9%)。临床表现:发作期最常见的临床表现为四肢肌肉无力(94.8%),其次是构音障碍、吞咽困难或两者兼有(86.2%),其他包括周围性面瘫、头晕呕吐、肢体麻木、共济失调等。辅助检查:56例患者进行周围神经系统检查,46例(82.1%)出现反射减退,35例(62.5%)有足部畸形,26 例(46.4%)出现四肢远端肌肉萎缩或无力。2 例(3.6%)周围神经系统检查未见异常。54 例患者在发病期行头颅MRI 检测,49 例(90.7%)可见脑室周围白质对称性长T1、长T2高信号,29 例(53.7%)出现胼胝体压部异常信号,8 例(14.8%)出现内囊后肢异常信号。48 例行神经电生理检查,除1 例[8]已报道的运动神经传导速度无异常,其余均下降。38 例进行了腰椎穿刺术,脑脊液结果:正常28例(73.7%),蛋白增高10例(26.3%),白细胞增高2 例(5.3%)。基因型分析:c.65G>A(p.Arg22Gln)报道了3 次,为出现次数最高的突变位点,c.425G>A(p.Arg142Gln)、c.490C>T(p.Arg164Trp),c.424C>T(p.Arg142Trp)重复报道两次,其余未见重复,45例患者符合X连锁的家族遗传特点,无男传男现象。治疗方案:30例报道了治疗方案,其中14 例(46.6%)接受了皮质类固醇和(或)静脉注射免疫球蛋白(intravenous immunoglobulin,IVIg)治疗,12例(40.0%)接受了对症和支持性治疗,4例(13.3%)未接受任何治疗。
X连锁遗传的CMT分为6型,CMTX1型占CMTX的90%[13],CMTX1、CMTX6 是X 连锁显性遗传,余为X 连锁隐性遗传[14]。其他的CMTX 分型未见发作性中枢神经系统障碍,但也可涉及中枢神经系统的功能障碍,如CMTX2型可见智力落后[15]。
CMTX1由GJB1基因突变引起,超过400个GJB1的突变位点已被发现[16],该基因负责编码缝隙连接蛋白Connexinx32(Cx32)的所有结构域[17]。研究表明,Cx32 缺失的小鼠在大约3 个月时出现进行性脱髓鞘神经病变、髓鞘变薄、洋葱球形成[18]。Cx32可分布在外周神经系统和中枢神经系统的髓鞘胶质细胞,参与缝隙连接的形成[19-20]。Cx32基因突变降低离子细胞通透性,抑制物质运输,引起细胞水肿,可能与头颅MRI显示异常相关。Cx32基因突变亦会引起缝隙连接蛋白组装障碍,抑制跨膜通道的形成或减小管腔直径,或对酸化诱导的关闭更敏感[21-23],高代谢、缺氧等诱发因素的出现往往会加速上述病理过程。
CMTX1患者的典型临床特征除具有经典的CMT的远端肌肉无力、萎缩、感觉障碍及反射减退或消失,也有少数患者出现发作性中枢神经系统表现,该症状多发生在儿童早期[24],常见有四肢无力、构音障碍、吞咽困难等,这类发作性症状通常持续几小时到几周不等。本例患者除表现为上述症状外还表现出轻度共济失调,该症状在24 h 内完全缓解。其他发作性的中枢神经系统症状可见脑神经损伤表现,如听力下降、面瘫、运动性失语、眩晕、呼吸困难、强直痉挛、手震颤、嗜睡等[25-26]。
MRI的改变通常在DWI(弥散加权序列)和T2加权序列可见,往往优先累及脑室周围白质和胼胝体压部,常对称分布[27],内囊和小脑脚也可出现异常信号[28]。磁共振成像的变化需要数天至数月才能恢复正常,通常滞后于症状恢复,既往研究认为恢复时间9 d~2 a[6]。目前普遍认为异常的头颅MRI 信号是可逆性的,可能与少突胶质细胞间的缝隙连接功能紊乱、髓内水肿有关[6]。因此,及时进行磁共振检查并动态随访是监测CMTX1 神经系统发作性症状伴白质病变的重要检查方法[29]。
CMTX1神经电生理显示广泛的周围神经传导速度下降35~45 m/s,属于中等传导速度减慢[30],同时出现动作电位波幅下降,男性下降更明显,提示周围神经轴索损伤伴脱髓鞘。TIAN 等[6]总结发现,当患者具有典型临床表现时,正中神经运动传导速度下降25~45 m/s,可支持CMTX1 的诊断。神经病理中既能见到髓球形成等轴索变性,又可见慢性脱髓鞘的“洋葱球”样病理改变,病理损害可随年龄增长而逐渐加重[31]。1例CMTX1病例报道仅发现脑干听觉诱发电位异常[12]。脑脊液检测仅发现少数患者出现轻度蛋白增高。
该病确诊需要GJB1基因的基因检测,同时结合家族史及家系共分离遗传学分析予以验证。伴发作性中枢神经系统障碍的CMTX1 患者的鉴别诊断包括:(1)基于影像学的脑白质病变,需与伴胼胝体压部可逆性病变的轻度脑炎/脑病、中毒、肾上腺脑白质营养不良(adrenoleukodystrophy, ALD)[32]等相鉴别。(2)发作性神经功能障碍的疾病表现形式需与短暂性脑缺血发作(transient ischemic atack, TIA)[33]、线粒体脑病伴乳酸酸中毒和卒中样发作(mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes, MELAS)[34]、急性播散性脑脊髓炎(acute disseminated encephalomyelitis,ADEM)[35]、周期性瘫痪等鉴别。本例患者第1 次发作时诊断为ADEM,启用免疫治疗后症状缓解,考虑为自然的病程缓解,与治疗无关。因此,有发作性中枢神经系统障碍的年轻患者,如果同时发现马蹄足、双下肢纤细,应考虑该病的可能。
CMTX1 发作时以支持治疗[36]为主,症状缓解后侧重于周围神经病的处理,包括物理治疗和矫形术的使用[36]。TIAN及其研究团队[6]总结病例时发现有较多的患者使用免疫球蛋白和激素治疗,包括本病例第1次发作经免疫治疗后症状缓解,均为自然的病程缓解,与治疗无关,因此不推荐免疫治疗。针对异常GJB1的基因替代治疗[38-39]很可能成为未来的有效治疗方案。
总之,对于有发作性中枢神经系统障碍的症状和头颅异常白质信号的年轻患者,应考虑CMTX1诊断可能性,询问家族遗传史并及时进行详细的体格及神经系统检查、颅脑MRI及电生理检测,包括遗传学分析,对于明确诊断和鉴别诊断十分必要。
