肋骨分叉-基底细胞痣-颌骨囊肿综合征两家系基因序列变异分析
2023-07-06彭笑陈默王栋韩瑞高廷益刘亮刘畅张凯
彭笑,陈默,王栋,韩瑞,高廷益,刘亮,刘畅,张凯
蚌埠医学院第一附属医院口腔科,安徽 蚌埠 233000
BCNS是一种常染色体显性遗传性疾病,最初在1894年由Jarisch描述。1960年,Gorlin和Goltz将BCNS归类为一种独特的综合征并提出“多发性痣样基底细胞癌、颌骨囊肿、分叉肋综合征”的概念[1]。BCNS在临床上有100多种疾病表型,主要包括多发性颌骨囊肿、面部或躯干多发性基底细胞痣(癌)、外/中胚层发育障碍、手掌和足底点状凹陷、脊柱发育异常等,此外还可伴有神经、生殖、皮肤等主要系统的病变[2-3]。BCNS的发病率为1/256 000~1/50 000,男女比例约为3∶1[4-5]。临床上,患者常因牙齿疼痛、口腔瘘管、颌面部肿大等主诉就诊,因此口腔科医生对于该疾病的认识和早期诊断十分重要。本文通过两例BCNS先证者及其家系相关成员的基因突变序列检测,在明确已知致病基因PTCH1的基础上,进一步探讨不同先证者及家系PTCH1基因突变位点的差异,并从分子遗传学角度揭示疾病遗传来源,以期为该疾病的早期诊断及治疗提供有益参考。研究方案遵守《赫尔辛基宣言》进行,并通过蚌埠医学院第一附属医院伦理委员会审查(2014010),先证者及其家系成员均签署知情同意书。
1 对象与方法
1.1 对 象
先证者1男性,14岁,于2021年8月31日以“X线摄片发现下颌骨囊肿一月余”为主诉就诊于蚌埠医学院第一附属医院口腔科。其母亲曾患颌骨囊肿。先证者1幼年曾于蚌埠医学院第一附属医院行“双侧唇裂修复术”和“腭裂修复术”。入院后临床检查结果:①智力低下;②前额宽大隆突,内眦距及瞳孔距增宽;③双侧下颌骨可触及膨隆,扪之有乒乓球样感;切牙孔至软腭有一垂直疤痕,左上切牙及侧切牙缺失,左上乳侧切牙及乳尖牙滞留,左上乳尖牙根尖部有一瘘管,按压疼痛并可见脓性分泌物渗出;④胸部X线摄片检查示右侧第3肋增粗、分叉;⑤头颅CT检查脑实质未见明显异常,小脑幕及大脑镰多发钙化灶;⑥牙齿全景片及锥形束CT检查显示多发性颌骨囊肿。见图1。先证者1颌骨囊肿开窗减压联合刮治术后病理检查(HE染色)可见鳞状上皮伴角化,局部间质内慢性炎症细胞浸润伴出血;镜下可见大量浆细胞及少量梭形细胞,符合角化囊肿改变,见图2。术后随访X线摄片检查显示原病灶处骨质生长状况良好,伤口无感染溢脓,至今未见复发征象。

图1 先证者1临床表现Figure 1 Clinical manifestations of proband 1
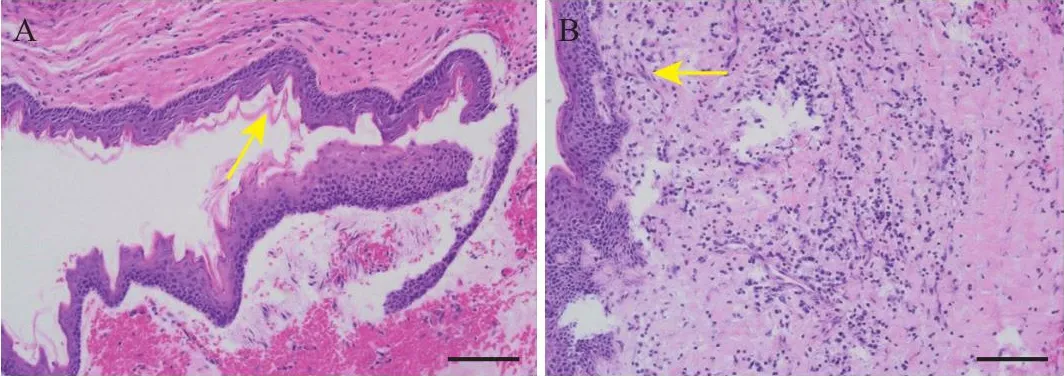
图2 先证者1颌骨囊肿病理检查结果(HE染色)Figure 2 Pathological examination results of proband 1 jaw cyst(HE staining)
先证者2男性,15岁,于2022年3月2日以“X线摄片发现上下颌骨多发性低密度影像”为主诉就诊于蚌埠医学院第一附属医院口腔科。其母亲曾患颌骨囊肿。入院后临床检查结果:①智力正常;②前额宽大隆突,内眦距及瞳孔距增宽;③左下第二磨牙、右下第一前磨牙、第二前磨牙及第二磨牙萌出,右下乳尖牙及第一乳磨牙滞留;④胸部X线摄片检查显示右侧第3、4后肋分叉,右侧胸廓略塌陷;⑤头颅CT结果提示小脑幕及大脑镰多发钙化灶,幕上脑室略扩张,左侧上颌窦炎;⑥牙齿全景片及锥形束CT检查显示多发性颌骨囊肿。见图3。先证者2行颌骨囊肿开窗减压联合刮治术后病理检查(HE染色)提示囊肿内衬复层鳞状上皮,局部伴感染,符合角化囊肿表现,见图4。术后随访X线摄片检查显示囊腔逐渐缩小,周围骨质生长状况良好,至今未见复发。

图3 先证者2的临床表现Figure 3 Clinical manifestations of proband 2

图4 先证者2颌骨囊肿病理检查结果(HE染色)Figure 4 Pathological examination results of proband 2 jaw cyst (HE staining)
1.2 仪器和软件
全血基因组DNA提取试剂盒Qiagen61104为美国Qiagen公司产品;Biorupter非接触式全自动超声破碎仪为比利时Diagenode公司产品;IGTTMFast Library Prep Kitv 2.0、AIExome Enrichment Kit V1为北京艾吉泰康生物科技有限公司产品;高通量测序系统Illumina NovSeq 6000为美国Illumina公司产品;SureSelect人全外显子探诊试剂盒V6为美国Agilent公司产品;Burrows Wheeler Alignment Tool(BWA)软件来源于http://sourceforge.net/projects/bio-bwa;Genome Analysis Toolkit(GATK)分析软件为美国Broad Institute公司开发;引物设计软件Snapgene为美国Insightful Science公司开发。
1.3 高通量测序验证基因突变
设计涵盖PTCH1基因编码区外显子的引物,对先证者及其家系成员DNA进行PCR扩增。其中先证者1的引物为PTCH1~15(5′→3′)正向TAGCCACCGTCTATCCAA、反向CCGAATATCCA GCACTTACT,先证者2的引物为PTCH1~10(5′→3′)正向CGGACAGC ATCAAGAGAC、反向GGTGGAGGGA AACATTAGAA,退火温度均为60 ℃。搜索HGMD及PubMed数据库以确定是否为新发突变;并依据ACMG最新基因变异数据解读指南对突变的致病性进行分析。
2 结果
2.1 两例先证者致病基因PTCH1突变位点检测
先证者1:PTCH1基因功能区域第2541号核苷酸由C突变为A,导致从第847号酪氨酸开始氨基酸编码终止,即c.C2541C>A(p.Y847X),为杂合截短突变(图5),符合常染色体显性遗传规律。先证者2:PTCH1基因功能区域第1501号核苷酸由C突变为T,导致从第501号谷氨酰胺开始氨基酸编码终止,即c.C1501C>T(p.Q501X),为杂合截短突变(图5),符合常染色体显性遗传规律。

图5 先证者及家系基因测序结果Figure 5 Gene sequence analysis of proband and family
2.2 两例先证者及家系序列遗传分析
两例先证者家系均检出受检者母亲DNA中存在PTCH1基因位点的杂合突变。根据ACMG指南,该突变可评级为致病:PVS1+PM2+PP4。PVS1:存在丧失功能致病机制的基因发生截短突变;PM2:该位点的人群频率无收录;PP4:变异携带者的表型或家族史与疾病高度符合。两例先证者及家系的树状图分析见图6。

图6 先证者及家系图分析Figure 6 Genetic map analysis of proband and family
2.3 其他基因突变序列
先证者1除PTCH1基因突变外,还检出3p25.3号染色体上的FANCD2基因位点的c.C2141T(p.P714L)和c.G3343A(p.V1115I)杂合突变,推测与智力低下临床表型有关。
3 讨论
BCNS可通过患者临床特征进行诊断[6]。虽然没有明确的基因型/表型相关性,但临床标准往往与基因突变的识别相匹配。由BCNS国际学术研讨会发布的诊断标准共识声明[7]可知,诊断BCNS的临床条件包括:①两项主要诊断标准;②一项主要诊断标准和两项次要诊断标准。其中主要诊断标准为:①多发基底细胞癌,20岁以下的单个基底细胞癌或10个以上的基底细胞痣;②病理学检查证实为牙源性角化囊肿;③多发性掌跖特殊凹陷;④颅内片层钙化或20岁以下的早期钙化;⑤家族中一级亲属患有BCNS。次要诊断标准为:正常身高发育完成后的巨头畸形、先天性颌面部畸形(唇腭裂)、额部隆起和中重度的眶距过宽、胸部畸形、脊椎异常、手脚畸形、先天性白内障、青光眼及子宫纤维瘤等。目前其发病机制尚不清楚,多数学者认为遗传和免疫是疾病发生及进展的主要原因[8-9]。
目前,分子生物学诊断仍是BCNS诊断的金标准[10]。BCNS是由果蝇patched-gene-1的人类同源基因PTCH1突变引起,在不同组织中具有不同的功能特性和表达模式。PTCH1定位于9q22.3号染色体上,约含24个外显子,编码1447个氨基酸的跨膜糖蛋白[11]。但有研究显示,突变位点与临床表型无关[12]。PTCH属于一种肿瘤抑制基因,主要通过诱导细胞凋亡抑制肿瘤形成,其突变可以激活Hedgehog信号通路,该通路在毛囊发育过程中可正常表达于表皮基底膜和间充质中[13]。研究证明,该通路异常可导致细胞无限增殖,临床上与一系列肿瘤发生和畸形相关[14]。BCNS患者中存在PTCH1的移码、删除、剪接位点和错义突变。此外,由9q22.3片段缺失引起的PTCH1单倍不足是导致BCNS综合征的原因[15-16]。本文资料两例BCNS先证者中,先证者1智力低下,PTCH1基因功能区域第2541号核苷酸由C突变为A,导致从第847号酪氨酸开始氨基酸编码终止;还存在FANCD2突变,核苷酸变化为c.C2141T和c.G3343A,氨基酸变化为p.P714L和p.V1115I。FANCD2也称为范可尼血D2蛋白,是DNA损伤修复相关的核蛋白;FANCD2基因有44个外显子,分别编码FANCD2-S和FANCD2-L两种长度包含1451个氨基酸残基的蛋白质,可修复DNA损伤,稳定染色体[17]。研究显示,染色体异常是导致儿童生长发育迟缓、智力低下、运动行为发育障碍及多发畸形的常见原因之一[18]。近年研究显示,铁死亡为细胞的程序性死亡方式,FANCD2可以保护骨髓基质细胞免受铁死亡所致的损伤,且有研究证实大鼠脑组织中发生铁死亡可能与FANCD2表达差异有关[19-20],但机制尚未明确。另有研究显示,BCNS患者中9q22.3号染色体的大量缺失可能是导致严重神经系统障碍如智力低下的原因[21-22]。先证者2智力发育正常,其PTCH1基因功能区域第1501号核苷酸由C突变为T,导致从第501号谷氨酰胺开始氨基酸编码终止,未发现FANCD2基因突变。智力低下是否与FANCD2基因突变有关仍有待进一步研究。
BCNS患者应避免暴露于紫外线及电离辐射区,以降低基底细胞癌的发生风险。因此,临床医生应熟悉并掌握该疾病的临床特征,做到正确评估和早期诊断,这对于BCNS患者的预后至关重要。本文资料两例先证者的母亲均为30岁以上X线摄片时发现颌骨囊肿,后确诊为BCNS,因此行家族筛查及产前基因检测是十分有必要的。建议对于临床接诊时遇到多发性颌骨囊肿的患者,应首先给予体格检查和胸部X线摄片、头颅CT等辅助检查,在明确相关器官异常的基础上行BCNS致病基因检测,若确诊则应对其家系成员行基因对比分析,以便进行后代生育指导和家族遗传来源、疾病发病概率分析。此外,针对BCNS患者中颌骨囊肿这一疾病表型,多数患者为上、下颌骨多发性囊肿,就诊时囊肿体积较大、骨质破坏严重,若直接行刮治术不仅容易损伤牙齿、下牙槽神经管、上颌窦等重要解剖结构及其功能,还会因囊肿数较多增加术中为打开囊腔而去除的骨窗数,从而使颌骨抗力降低,相比单个囊肿更易导致病理性骨折的发生。因此建议对于BCNS患者的颌骨囊肿,可先行开窗减压,待骨质沉积量增加或病灶已远离相关重要解剖结构时再行内镜辅助下刮治术,术中磨出囊腔周围1~2 mm骨质,术后嘱患者定期随访以观察囊腔的变化情况及有无复发。BCNS常导致先天性颅颌面缺陷及严重的错颌畸形[23],颌骨囊肿常导致邻近牙齿移位,BCNS患者囊肿术后应进行后续正畸治疗,从而建立正确、稳定的咬合关系,提高患者远期生存质量。
本研究采用高通量测序基因筛查手段对BCNS家系行基因检测,该方法具有高效性和特异性,能够准确检测出致病基因序列及位点变化,并可针对性揭示家系遗传来源及可能存在的其他类型基因突变。临床上对于BCNS应尽可能早期行基因检测明确分子生物学病因及家系遗传情况,以此作为疾病来源标准及生育相关指导,通过多学科联合治疗模式对患者可能存在的多种疾病表型进行早期治疗,这对于BCNS患者的整体诊疗及预后具有积极意义。
志谢研究得到安徽省高校学科(专业)拔尖人才学术资助项目(gxbjZD2021058)支持
AcknowledgmentsThis work was supported by Anhui Province University Discipline (Major) Top Talent Academic Support Project (gxbjZD2021058)
利益冲突所有作者均声明不存在利益冲突
Conflict of InterestsThe authors declare that there is no conflict of interests
©The author(s) 2023.This is an open access article under the CC BY-NC-ND 4.0 License (https://creativecommons.org/licenses/by-nc-nd/4.0/)
