探索m6A 相关lncRNAs 预测胃腺癌患者总体生存率
2023-07-03潘辉姚文霞梁敏周新科
潘辉,姚文霞,梁敏,周新科
510700 广州,广州医科大学附属第五医院 前沿医学交叉研究中心(潘辉、姚文霞),肿瘤科(梁敏、周新科)
胃癌每年估计有超过100 万新发病例,是全球第五大恶性肿瘤。近期发布的2016 年中国恶性肿瘤流行情况分析显示,胃癌居我国恶性肿瘤发病第三位[1]。由于诊断手段不足,胃癌被诊断时,大部分患者处于晚期,患者治疗效果不佳,死亡率较高,全球范围看,胃癌排在癌症相关死亡的第三位[2]。胃腺癌是最常见的胃癌类型,约占所有胃癌的90% ~ 95%。
N6-甲基腺苷(N6-methyladenosine,m6A)是真核细胞中最丰富的RNA 修饰,在RNA 加工、转运和稳定性等各种生物过程和mRNA 代谢中起着至关重要的作用[3-5]。m6A 甲基化修饰调控因子包括甲基化转移酶、甲基化阅读蛋白和去甲基化酶,它们分别被称为写入器、读取器和擦除器。此外,m6A 修饰是可逆的RNA 表观遗传过程[6]。RNA 结构的改变可以影响各种细胞过程。因此,m6A 调控长链非编码RNA 的作用可能对癌细胞的增殖和迁移至关重要[7-8]。
最近的研究表明m6A 修饰调控肿瘤发生和肿瘤发展。例如,受m6A 修饰影响的FEZF1-AS1调控ITGA11/miR-516b-5p 轴,最终在非小细胞肺癌中上调[9]。此外,m6A 甲基转移酶样3 诱导的lncRNA ABHD11-AS1 在非小细胞肺癌中表达上调,其异位表达与非小细胞肺癌患者预后不良密切相关[10]。近年来,多项生物信息学研究表明,m6A调控因子的失调参与了胃腺癌的发生[11-13]。了解m6A 调控的长链非编码RNA(long non-coding RNAs,lncRNAs)在胃腺癌发生中的机制可能有助于预测疾病的预后。本研究从15 901 个lncRNAs中筛选出12 个m6A 相关lncRNAs 构建风险模型,这些lncRNAs 被确定为影响胃腺癌患者预后的独立预测因子。此外,本研究还探讨了风险评分因子与肿瘤微环境细胞浸润的关系。最后,构建了一个列线图用来个体化预测胃腺癌患者的总生存期,研究思路如图1 所示。

图1 研究思路Figure 1.Study Design
1 材料与方法
1.1 胃腺癌患者转录组信息的获取
本研究从TCGA(https://cancergenome.nih.gov/)数据库中获取了胃腺癌患者的RNA 序列转录组数据和相关临床信息。为减少研究分析的统计偏倚,排除了总生存期缺失的部分胃腺癌患者。
1.2 m6A RNA 甲基化调控因子及相关lncRNAs的选择
基于TCGA 数据库中获得了lncRNAs 和m6A修饰调控因子的表达谱数据。根据以往的研究,从TCGA 中检索了21 个m6A 修饰调控因子的表达矩阵[14-16],包括甲基化修饰转移酶(METTL3、METTL14、METTL16、VIRMA、RBM15、RBM15B、ZC3H13、WTAP),甲基化修饰识别蛋白(IGF2BP1、YTHDC1、YTHDC2、YTHDF1、YTHDF2、YTHDF3、HNRNPA2B1、HNRNPC、ELAVL1、FMR1、LRPPRC)和去甲基化酶(ALKBH5、FTO)的表达数据。根据GENECODE 对TCGA 数据基因类型的定义,挑选出归属于lncRNAs 类。经过筛选,研究数据中入选的lncRNAs 共15 901 个。使用R 语言的“limma”包对15 901 个lncRNAs 按肿瘤组织和正常组织分组进行差异表达分析。以logFC 绝对值的平均值±2 个标准差为“cutoff”值[mean(|logFC|)±2SD(|logFC|)],P值小于0.05 为阈值划分表达量差异是否有统计学意义。
1.3 风险评分模型的建立和验证
结合数据集中胃腺癌患者生存信息,采用单因素Cox 回归分析,从TCGA 数据集中的347 个与m6A 相关lncRNAs 中筛选出与预后相关的基因67个(P< 0.05)。为进一步确定影响患者的独立预后因素,使用R 包“glmnet”对67 个入选lncRNAs 进行LASSO Cox 回归分析。我们发现TCGA 数据集中12 个m6A 相关的lncRNAs 与胃腺癌患者的总生存期明显相关。采用多因素Cox 回归分析12 个m6A相关的lncRNAs,最终建立起m6A 相关的lncRNAs风险评分模型。利用以下公式计算风险评分:
riskScores = coef (lncRNA1)×expr (lncRNA1)+ coef (lncRNA2)×expr (lncRNA2) +…+ coef (lncRNAn)×expr (lncRNAn)。
其中coef 表示风险系数,coef (lncRNAn)为与生存相关的lncRNA 的系数,expr (lncRNAn)为lncRNA 的表达量。使用“surv_cutpoint”包寻找危险度评分的最佳截点,将患者分为低危险度组和高危险度组。
1.4 探索风险评分与肿瘤微环境细胞浸润的关系
整理出与肿瘤免疫相关的基因集,采用m6A相关lncRNAs 风险评分模型对基因进行分组。对379 例胃腺癌肿瘤标本中免疫细胞的富集水平和活性进行分析,展示风险评分与免疫相关基因的相关性。接下来使用ESTIMATE 算法计算胃腺癌的肿瘤纯度。通过风险评分分组,计算不同分组的“estimate score”,进而反应出不同分组的肿瘤纯度状况。
1.5 主成分分析(principal component analysis,PCA)和Kaplan-Meier 生存分析
根据12 个m6A 相关lncRNAs 的表达模式,采用PCA 对整个基因表达谱、21 个m6A 调控因子、12 个m6A 相关lncRNAs 的高维数据进行有效降维、模型识别和分组可视化。同时采用Kaplan-Meier 生存分析来评估高危组和低危组之间总生存期情况,使用“survMiner”和“survival”R 包绘制生存曲线。
1.6 预测列线图的构建与评价
综合胃腺癌患者的其他临床特征(性别、年龄、TNM 分期、T 分期、N 分期、M 分期、病理分化程度)进行单因素Cox 回归分析和多因素Cox 回归分析,以检验上述因素是否是影响预后的独立因素。
基于独立的预后影响因素建立列线图,了解个性化预测患者1 年、3 年和5 年的生存概率。并检测生存预测模型的预测能力,基于Hosmer-Lemeshow 检验修正曲线来说明实际结果与模型预测结果的一致性。
2 结 果
2.1 胃腺癌患者m6A 相关lncRNAs 的鉴定
本研究从TCGA 数据库中提取了21 个m6A调控因子和15 901 个lncRNAs 基因的表达量数据。使用“limma”包对lncRNAs 基因的表达量进行差异分析。分析结果发现共有344 个lncRNAs 表达量下调,414 个lncRNAs 表达量上调,如图2 和图3 所示。

图2 肿瘤组织与正常组织lncRNAs 表达量情况Figure 2.LncRNAs Expressions in Tumor and Normal Tissues
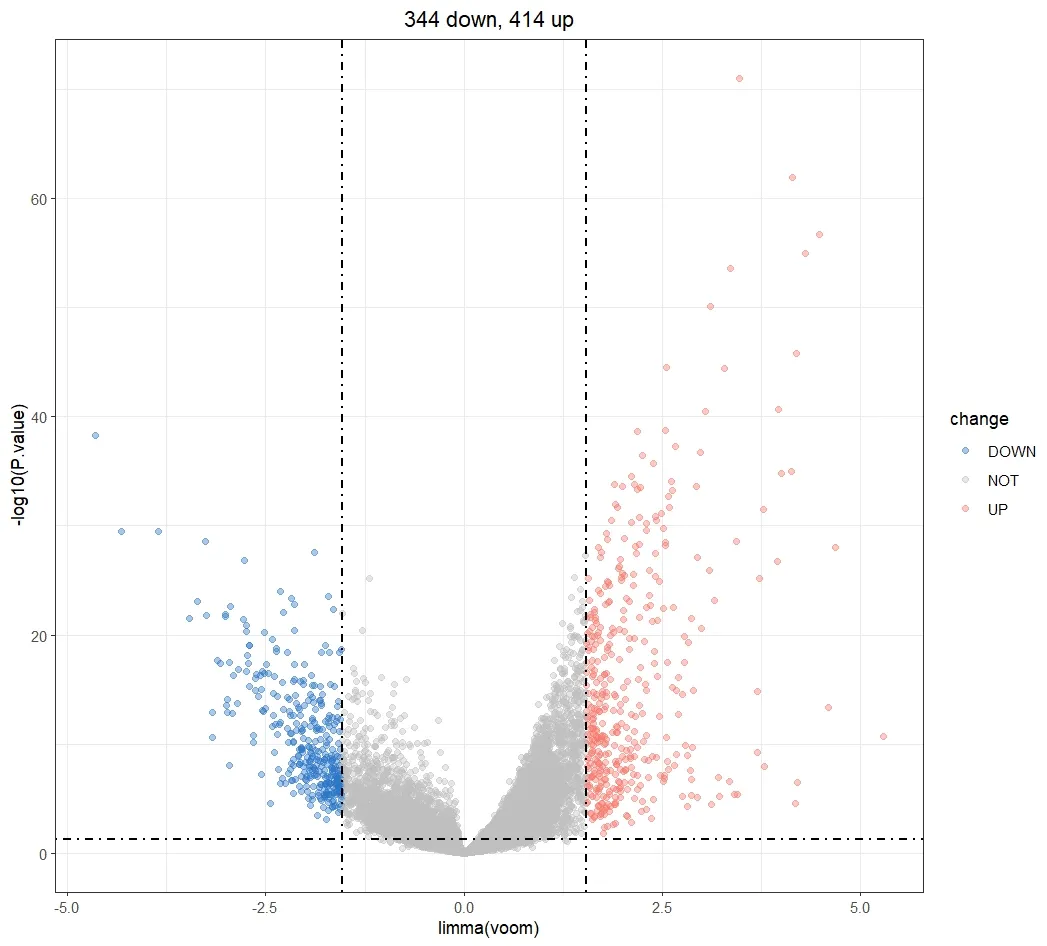
图3 差异基因火山图Figure 3. Volcano Plot of Differential Genes
将m6A 调控因子相关的lncRNAs 定义为两者Pearson 相关系数大于0.3,且P值小于0.001(|Pearsonr| > 0.3,P< 0.001)。共鉴定出1 250 组m6a 相关lncRNAs,其中共包含347 个lncRNAs,使用图4 中的Sankey 图对m6A-lncRNAs 共表达网络进行可视化,图5 展示相关系数前50 的m6A-lncRNAs 关系网络。

图4 21 个m6A 调控因子和m6A 相关lncRNAs 的Sankey 关系图Figure 4.Sankey Diagram of the Relationship between m6A Regulators and m6A-Related LncRNAs

图5 21 个m6A 调控因子和m6A 相关lncRNAs 的关系系数图Figure 5.Relationship between m6A Regulators and m6A-Related LncRNAs
2.2 胃腺癌患者m6A 相关lncRNAs 风险评分模型的构建与验证
本研究使用单因素Cox 回归分析,从TCGA训练集中的1 250 组m6A-lncRNAs 中筛选出与患者预后相关的lncRNAs。TCGA 数据集中67 个m6A 相关的lncRNAs 与患者的总生存期显著相关(P< 0.05),用森林图的形式随机展示了30 个lncRNAs 的HR及95%CI(图6)。

图6 单因素Cox 回归分析随机展示30 个m6A 相关lncRNAs 与患者预后的关系Figure 6.Relationship between m6A-Associated LncRNAs and Prognosis by Univariate Cox Regression Analysis
进一步采用LASSO Cox 回归分析方法对上述67 个lncRNAs 进行选择。如图7 和图8 所示,虚线垂线表示具有最小分段似然偏差的lambda 值。因此,我们选择12 个m6A 相关的lncRNAs(包括AL589743.3、AC007128.2、AC130324.2、AP001107.5、AC090809.1、AC109782.1、LINC01537、ADAMTS9-AS1、AL391095.1、AC084880.3、AC012055.1、AL359704.2)进行后续的多因素分析。

图7 LASSO 回归分析挑选出合适的lambda 值Figure 7. Suitable Lambda Values Selected by LASSO Regression Analysis

图8 LASSO 回归分析筛选出12 个lncRNAsFigure 8.12 LncRNAs Screened by LASSO Regression Analysis
接下来,我们使用多因素Cox 危险比回归分析证实12 个m6A 相关lncRNAs 是影响胃腺癌患者预后独立的因素。基于12 个lncRNAs 构建风险评估模型,评估胃腺癌患者的预后风险,使用ROC 曲线评价模型的预测精确性,模型预测能力的AUC 值为0.68,能较好地预测患者的生存状况,具体如图9 所示。

图9 ROC 曲线评价模型的预测精确性,模型预测能力的AUC 值为0.68Figure 9.Accuracy of the Model Evaluated by ROC Curve with the AUC of 0.68
使用“survminer”包中的“surv_cutpoint”函数测算预后风险等级的截点(cutpoint = 1.2),将胃腺癌患者样本分为低危风险组和高危风险组,其中低危组患者207 例,高危组患者145 例,如图10 所示。低风险组和高风险组之间的风险等级分布,两个不同风险组患者的生存状态和生存时间,以及每位患者12 个lncRNA 的相对表达情况均在图11 中展示。由图可见,高风险组的风险评分等级较高,患者死亡人数占比较多,以及相关lncRNAs的表达量较高。生存分析表明,低危组的总生存期长于高危组(P< 0.001),如图12。

图10 显示风险评分的分组截点,截点值为1.2Figure 10.Distribution of Risk Scores with the Cutpoint Value of 1.2

图11 不同分组患者的风险等级、生存状况及lncRNAs 的表达量Figure 11.Risk Scores, Survival and Expression Levels of LncRNAs in Different Groups

图12 低风险组患者总生存期明显优于高风险组Figure 12.Overall Survival in the Low-Risk Group Was Significantly Better than That in the High-Risk Group
为进一步评估风险评估模型对患者预后的预测作用,本研究将患者根据临床病理特征分层,如按性别、年龄、分期、肿瘤分化程度划分亚组,比较各亚组中低危组和高危组患者总生存期情况,结果显示低危组总生存期依然优于高危组(图13) 。

图13 不同亚组中低风险组患者总生存期明显优于高风险组Figure 13.Overall Survival in Low-Risk Groups Were Significantly Better than Those in High-Risk Groups
经过初步的验证,多因素Cox 分析构建12 个m6A 相关lncRNAs 风险评估模型可以评估胃腺癌患者的预后风险。
2.3 PCA 进一步验证风险评估模型的分组能力
基于风险评估模型分类,分别对全基因表达谱、所有lncRNAs 基因集、21 个m6A 调控因子和12 个m6A 相关lncRNA 表达量进行主成分分析以测试低风险组和高风险组之间的差异。图14 显示,高风险对于低风险人群的分布相对分散。同时,低风险组和高风险组具有不同的分布。通过主成分分析进一步验证了12 个m6A 相关lncRNA 风险模型可以有效区分胃腺癌患者分类。

图14 基于不同风险分组对全转录组基因表达谱(A)、全lncRNAs 基因(B)、m6A 调控因子(C)和12 个m6A 相关lncRNAs(D)进行主成分分析Figure 14.Principal Component Analysis was Performed for Transcriptome-Wide Gene Expression Profiles (A), the Whole LncRNAs (B), m6A Modulators (C) and 12 m6A-Related LncRNAs (D) in Different Groups
2.4 用m6A 相关lncRNAs 模型评价肿瘤微环境免疫细胞浸润的情况
为了探索m6A 相关lncRNAs 模型与肿瘤免疫微环境(tumor immune microenvironment, TIME)免疫细胞浸润中相互作用,本研究评估了不同风险分组中TIME 细胞浸润的情况。Charoentong 等[17]2017 年发表于《Cell Reports》的研究论文中,汇总了28 种免疫细胞的基因集。本研究根据此基因集,整理出免疫细胞的表达谱数据,进而分析不同风险组免疫细胞富集情况。如图15 所示, effector memory CD4 T cell、mast cell、activated CD4 T cell、immature dendritic cell、eosinophil、plasmacytoid dendritic cell、macrophage、activated B cell、T follicular helper cell、type 1 T helper cell、memory B cell、monocyte、CD56dim natural killer cell、 immature B cell、central memory CD8 T cell、activated CD8 t cell、type 17 T helper cell、central memory CD4 T cell 共18 个免疫细胞组免疫指标表达水平低风险组和高风险存在显著差异。其中在低风险组中,activated CD4 T cell、CD56dim natural killer cell、activated CD8 T cell、type 17 T helper cell 4 种免疫细胞显著富集,其余14 个免疫细胞组表达量高风险组显著富集。

图15 不同风险组中18 项免疫指标的表达上存在显著差异Figure 15.Significant Differences in the Expression Levels of 18 Immune Indicators in Different Risk Groups
为了进一步确定低风险组和高风险组之间TIME 细胞浸润模式是否存在差异,我们使用PCA算法对28 个TIME 浸润细胞进行降维。我们观察到两个独立的TIME 细胞群(图16),表明胃腺癌中TIME 细胞浸润模式的变化可能介导胃腺癌转移、免疫逃逸和对免疫疗法的抵抗。

图16 对两个相对独立的群体的28 个TIME 浸润细胞的主成分分析显示不同风险组之间的TIME 免疫细胞浸润情况存在差异Figure 16.Principal Component Analysis of TIME Infiltrating Cells in Two Relatively Independent Populations Showed Differences in TIME Cell Infiltration between Different Risk Groups
我们使用估计算法来评估胃腺癌微环境中的免疫和基质活性,发现高风险组中的基质活性比低风险组的显著增高。为了探讨关键lncRNAs 与TIME浸润细胞之间的关系,我们将关键分子与TIME 浸润细胞进行了关联。Spearman 相关性分析显示, 12个lncRNAs 分子与TIME 浸润细胞之间存在显著的相关性(图17)。上述结果揭示,m6A 相关lncRNA风险模型可以用于评估TIME。

图17 12 个lncRNAs 与每种TIME 浸润细胞类型之间的相关性Figure 17.Correlations between 12 LncRNAs and TIME Infiltrating Cells
2.5 m6A 相关lncRNAs 风险模型影响预后的评价及与临床特征的关系
本研究进行了单因素和多因素Cox 回归分析,以评估12 个m6A 相关lncRNAs 构建的风险评分模型是否具有影响胃腺癌预后的独立特征。在单因素Cox 回归分析中,风险评分的HR和95%CI分别为3 和2.2~4.3(P< 0.001)。在多因素Cox 回归分析中,HR为3.5,95%CI为2.46~4.99(P< 0.001)(图18),表明风险评分模型与患者预后密切相关。

图18 单因素和多因素Cox 回归分析均显示出风险评分模型是影响患者预后的独立因素(P < 0.001)Figure 18.Univariate and Multivariate Cox Regression Analyses Showed That the Risk Scoring Model Was an Independent Factor Affecting the Prognosis of Patients (P < 0.001)
本研究使用一致性指数和AUC 评估风险评分模型的效度和信度,风险等级预测1 年、3 年和5 年的AUC 值均高于0.70,表明风险评分模型对胃腺癌的预后预测结果的可靠性,如图19所示。

图19 风险评价模型预测患者预后能力的评价,AUC 值均高于0.70,显示出预测结果的可靠性Figure 19.AUC Values (> 0.70) Suggesting the Reliability of the Risk Scoring Model in Predicting Prognosis
2.6 预测列线图的构建与评价
构建包括风险等级和临床风险特征的列线图,以图形样式预测个体患者1 年、3 年和5 年的整体存活率。与其它临床因素相比,预后模型的风险等级在列线图中显示出主要的预测能力,具体见图20。预后模型的C-index 值为0.73,表示预测结果与实际观察到的结果相一致的概率较高,证明模型的预测能力可靠。通过判别能力和校准能力来评价列线图模型有效性,绘出胃腺癌1 年、3 年和5 年的标准曲线,标准曲线比较靠近对角线,表明列线图模型预测结果与 Kaplan-Meier 法结果一致性高,具体见图21。

图20 纳入风险评分的预测模型列线图,风险等级在列线图中显示出突出的预测能力Figure 20.A Nomogram for Predicting Overall Survival Probability of Patients
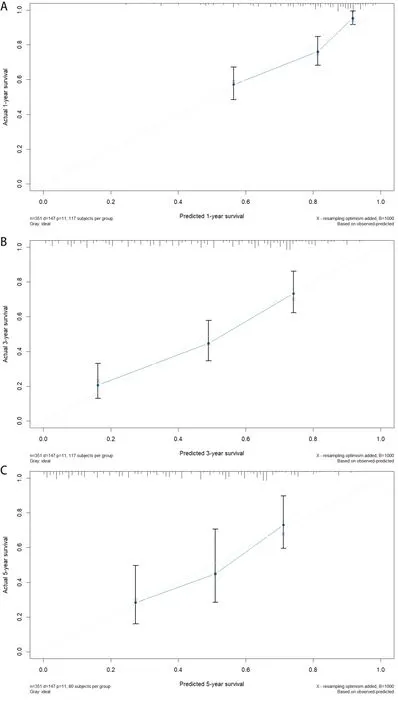
图21 预测模型1 年、3 年和5 年的标准曲线Figure 21.Calibration Plots of Model Predicting 1- (A), 3- (B), and 5-Year (C) Survival in the Complete Dataset
3 讨 论
胃腺癌作为胃癌最常见的亚型,大量医学研究者致力于胃腺癌的发生、发展和治疗的研究。m6A 是一种转录后修饰,可以影响RNA 的表达和功能。多项研究表明在癌症中m6A 的调节异常会导致体内信号传导通路的崩溃,加速肿瘤的发生和发展[18-21]。LncRNAs 是一种长度超过200 个核苷酸的非编码RNA,它的调节异常能够导致癌症的发生和进展。最近的研究表明,m6A修饰也能够影响lncRNAs 的表达和功能,进而对癌症的发生和发展产生影响[22-24]。LncRNAs可能将m6A 调控因子作为竞争性内源性RNA,影响肿瘤侵袭性进展。m6A 修饰可以通过向m6A 读取器蛋白提供结合位点来调节lncRNA功能。它还可以调节局部RNA 结构,使具体的RNA 结合蛋白进入周围的m6A 残基。再者m6A 可能影响lncRNA 和特异性DNA 之间的相互作用位点。如在胃癌中,一些lncRNA(如HOTAIR 和MALAT1)的m6A 修饰异常会导致它们在胃癌细胞中的过度表达,从而促进胃癌的发生和发展[25]。因此,m6A 甲基化修饰的调控是胃癌中调控lncRNA 的一个重要机制之一。
然而,关于胃腺癌涉及m6A 相关的lncRNA的生物学机制和预后生物标志物的研究仍不够充分。在本研究中,我们受到m6A 和lncRNAs 在胃腺癌中的作用的启发,试图构建一个基于m6A相关lncRNAs 的独立风险评分模型。从TCGA数据库的胃腺癌数据集中鉴定出差异基因,以此为基础,探索m6A 调控因子和差异lncRNAs 的相关性。发现347 个lncRNAs 与m6A 调控因子相关,用以探索其影响患者的预后作用。在探索两类基因的相关性时,相关系数r值 > 0.3,且P值小于0.001 的组共有1 250 组;而相关系数r值小于-0.3,且P值小于0.001,却没有一组。可揭示两类基因具有同向特点,如果某个m6A 调控因子是肿瘤的危险因素,那么与其相关的lncRNAs 也可能是该肿瘤的危险因素。
基于上述m6A 相关的lncRNAs,TCGA 数据集证实了67 个m6A 相关lncRNAs 的预后价值,并将其中12 个应用于构建m6A 相关性lncRNAs 风险评分模型(riskScore),以预测胃腺癌患者的总生存期。按风险评分高低将研究队列患者分为高危风险组(n= 145)和低危风险组(n= 207),并对此模型的预测患者预后的可靠性进行了多方面的验证。首先,分析了风险评分分组中患者生存状况分布的情况,可见高风险组患者死亡的比例远高于低风险组。其次,入选的12 个lncRNAs 的表达量在不同风险分组中存在显著差异。最后,分析了风险评分与患者临床信息的关系,在年龄、性别、病理分化程度和TNM临床分期中,我们都可以看到无论是在哪个亚组中,低危风险组患者的总生存期都要较高危风险组患者的长。
此外,本研究从TIME 免疫细胞浸润方面再次验证风险评分模型的区分能力。我们汇总了28 种免疫细胞的基因集,并整理出免疫细胞的表达谱数据,进而分析不同风险组免疫细胞富集情况[17]。18 种免疫细胞组免疫指标表达低风险组和高风险存在显著差异。其中低风险组中,activated CD4 T cell、CD56dim natural killer cell、activated CD8 T cell、type 17 T helper cell 4 个免疫细胞显著富集,其余14 个免疫细胞组表达量高风险组显著富集。我们使用估计算法来评估胃腺癌微环境中的免疫和基质活性,发现高风险组中的基质活性比低风险组的显著增高。为了探讨关键lncRNAs 与TIME浸润细胞之间的关系,我们将关键分子与TIME 浸润细胞进行了关联。Spearman 相关性分析显示,这些分子与TIME 浸润细胞之间存在显著的相关性。越来越多的证据表明,单个关键分子可以通过改变微环境免疫细胞浸润特征来诱导免疫耐受,并通过重塑微环境结构来避免免疫攻击[26-28]。本研究进一步探索lncRNAs 是否可以调节TIME 细胞的浸润水平,结果揭示了关键分子的表达与18 个TIME 细胞浸润之间的显著负相关,这证明风险评估模型与TIME 细胞浸润存在相关性。
多元Cox 回归分析表明,风险评分模型是胃腺癌患者关键危险因素。ROC 分析表明,以风险评分作为其中一项变量的Cox 回归模型在胃腺癌的生存预测方面表现出优异的敏感性和特异性。本研究建立了一个列线图用于个性化预测患者的总生存状况。在临床上,病理分化程度是胃腺癌预后的决定性因素。然而,同一分化阶段的胃腺癌患者的临床结果总是存在差异,这表明目前的分期系统在提供可靠的预测和反映胃腺癌异质性方面是不准确的。因此,应探索潜在的预测和治疗生物标志物,丰富现有的分期系统。本研究所建立的m6A 相关lncRNAs 模型为胃腺癌患者的预后预测提供了一种新方法。该结果也为未来对lncRNA-m6A 修饰的过程和机制的研究提供了见解。我们也意识到这项研究中的一些不足和局限性,如缺乏外部数据验证,并且m6A 相关lncRNAs 的生物学机制尚未完全阐明。因此,我们将重新收集临床样本并扩大样本量。此外,我们将尝试通过更多的外部实验来验证该模型的准确性,以进一步证实lncRNAs 的作用及其与m6A 相关基因的相互作用。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
