猪德尔塔冠状病毒病原学特征与致病机制研究进展
2023-07-01朱培英冉李艳杨莎莎施徐森李文贵
朱培英,冉李艳,杨莎莎,施徐森,李文贵
( 云南农业大学动物医学院,云南 昆明 650201 )
猪德尔塔冠状病毒(porcine deltacoronavirus,PDCoV)又称猪丁型冠状病毒,为套式病毒目(Niovirales)、冠状病毒科(Coronaviridae)、冠状病毒亚科(Coronavirinae)、德尔塔冠状病毒属(Deltacoronavirus)成员[1]。PDCoV 最早于2012年在香港的猪直肠拭子中检测到,2014年初在美国俄亥俄州猪场暴发,随后在加拿大、韩国、泰国、老挝、越南及中国大陆的养猪场中均有PDCoV 检出[2]。PDCoV 是引起猪群腹泻的重要病原之一,主要通过胃肠道传播并迅速蔓延。感染PDCoV 主要的临床表现是水样腹泻、呕吐和脱水;病变主要位于小肠,尤其是空肠和回肠,特点是肠壁变薄,肠道内充满气体和黄色液体,肠黏膜呈串状充血[3-4]。哺乳仔猪感染PDCoV 后死亡率高,生长猪和成年猪感染后死亡率低。
2021年,有研究发现3名患有急性未分化发热性疾病的海地儿童血浆样品中存在PDCoV,这是首次在人类体内发现该病毒,给动物和人类健康安全带来了巨大威胁[5]。由于PDCoV致病机制尚未明确,缺乏疫苗和特效药,成为当前养猪业疫病防控难点。本文从PDCoV 各蛋白出发,探讨该病毒如何通过细胞膜上的受体入侵细胞,维持自身增殖,诱导细胞凋亡,调节细胞自噬,并通过复杂的组合机制以及细胞分子和宿主蛋白参与致病过程,以期为PDCoV的防治方法和疫苗研发提供参考。
1 PDCoV病原学
PDCoV 是一种含囊膜的单股正链RNA 病毒,囊膜外有冠状棘突,病毒颗粒大小为60~180 nm。PDCoV基因组全长约25.4 kb,GC含量约43%[6],基因结构包含5'和3'非翻译区及至少7个开放性阅读框,分别编码多聚酶蛋白(polymerase protein)1a/1b、纤突蛋白(spike,S)、膜蛋白(envelope,E)、膜蛋白(membrance,M)、核衣壳蛋白(nucleocapsid,N)、非结构蛋白6(nonstructural protein 6,NS6)及非结构蛋白7(nonstructural protein 7,NS7)。目前对PDCoV 的结构蛋白、非结构蛋白以及辅助蛋白的功能只有初步的研究,仍需进一步探索。
1.1 PDCoV结构蛋白及功能
1.1.1 S蛋白
S 基因全长3 483 nt,编码1 161 个氨基酸。S 基因是PDCoV 纤突蛋白基因,是一种同源三聚体,每个单体分为两个区域,即S1和S2。S1蛋白是球形结构,包含两个独立的功能亚域N-末端(S1-NTD)、C-末端(S1-CTD)和相应的受体结合域(RBD)。S1可以与宿主细胞膜结合[7],S2是高度保守的螺旋,包括两个七肽重复区(HR1 和HR2)、一个跨膜区(TM)和一个融合肽区(FP)。S2的羧基末端构成了刺状蛋白的茎,参与病毒和宿主细胞膜的融合。S 基因变异较大,且变异速度较快,是唯一一个负责进入细胞的病毒膜蛋白,与靶细胞上的受体结合,之后介导病毒与细胞融合,导致机体患病[8]。
张家林[9]研究表明,在内吞体中CTSL和CTSB的切割下,S 蛋白活化暴露出S2 N 端的融合肽;融合肽插入内吞体膜,导致病毒囊膜与内吞体膜融合,病毒释放进入细胞,之后开始新一轮的复制增殖。此外,在细胞膜表面外源添加胰酶的处理下,S蛋白也被切割活化,导致病毒囊膜与细胞膜融合,病毒由此入侵细胞。Dong 等[10]研究发现,PDCoV的S蛋白与其他冠状病毒具有相同的功能,在病毒与宿主结合和产生中和抗体方面发挥重要作用,是冠状病毒的主要抗原蛋白,也是新疫苗研究的首选蛋白。
1.1.2 E蛋白
E 基因全长252 nt,编码84 个氨基酸,位于膜表面,是PDCoV基因组中最小的结构蛋白。目前对PDCoV E蛋白的研究报道很少,根据其他冠状病毒E蛋白的相关研究可知其含量很低,但对病毒的组装、出芽和细胞内运输具有一定作用,且这些作用可能与病毒的致病机制存在某些关联。病毒组装时,E 蛋白与M 蛋白相互作用形成复合体,辅助M蛋白进入病毒的包膜结构内[11]。
1.1.3 M蛋白
M 基因全长654 nt,编码218 个氨基酸,位于囊膜表面,是病毒囊膜表面相对丰富的跨膜蛋白,而且相对保守,可作为靶蛋白建立检测PDCoV 抗体的间接ELISA 方法[12]。M 蛋白在保护机体和介导疾病过程中发挥重要作用,主要参与营养物质的跨膜运输、形成新病毒颗粒的包膜和病毒颗粒释放。
1.1.4 N蛋白
N 基因全长1 026 nt,编码342 个氨基酸,可刺激机体产生免疫应答抗原蛋白。通过对比不同毒株N 蛋白氨基酸可知N蛋白高度保守(97.1%~100.0%)[13]。基于此,许多研究建立了针对PDCoV N 蛋白的检测方法。如朱小甫等[14]、黄海鑫[15]、苏红等[16]分别建立不同基于PDCoV N 基因的RT-nPCR 检测方法,对各地区猪群PDCoV 感染情况进行了流行病学调查。
1.2 PDCoV非结构蛋白和辅助蛋白功能
1.2.1 ORF1a、ORF1b蛋白
与其他冠状病毒类似,PDCoV 的ORF1a、ORF1b 可编码两个多聚蛋白(pp1a、pp1b),pp1a 和pp1b 可被蛋白酶切割水解加工产生15个非结构蛋白(NSP1~NSP15),一些非结构蛋白已被证明参与病毒的转录和复制。如NSP15 以二聚体发挥酶活的作用机制,能够特异性切割双链或单链核苷酸尿嘧啶,参与病毒复制过程,是病毒重要的毒力蛋白[17]。NSP3 编码木瓜样蛋白酶,而NSP5 编码3C 样蛋白酶,两者均在切割和加工多聚蛋白的过程中发挥作用[18]。NSP12是病毒负链RNA合成的过程中不可或缺的RNA聚合酶,NSP14 具有N7-甲基转移酶活性和核酸外切酶活性,两者共同参与了病毒蛋白水解与基因组RNA的复制。
1.2.2 辅助蛋白
尽管辅助蛋白对冠状病毒的复制并非必不可少,但其已被证明在机体的免疫调节、致病机制和毒力方面发挥重要作用。因此,鉴定和研究辅助蛋白可提供关于其在病毒生命周期中的具体功能的信息。Fang等[19]发现,辅助蛋白NS6 过表达通过与RIG-I/MDA5 相互作用显著抑制仙台病毒诱导的β 干扰素产生,以及转录因子IRF3 和NF-κB的激活。Co-IP竞争性试验结果也证实了NS6能够显著减弱RIG-I/MDA5 与dsRNA 的结合,表明PDCoV NS6 采用了一种新的免疫抑制策略,抑制RLRs 介导IFN-β 产生。上述结果表明,NS6是PDCoV 的一个重要毒力因子,在病毒复制和致病机制中发挥重要作用,因此缺乏辅助NS6蛋白的PDCoV可能是一个潜在的疫苗候选者。还有研究分析了与NS6相互作用的病毒编码蛋白和宿主蛋白,结果有助于进一步阐明NS6在病毒复制和致病机制中的作用,为PDCoV防控和药物靶点筛选提供理论依据。
此外,NS7 也是PDCoV 的辅助蛋白,Choi 等[20]研究发现,NS7 蛋白能够减少大鼠辅肌动蛋白α4(α-actinin-4,ACTN4)的表达。许多病毒操纵肌动蛋白进入宿主细胞,而ACTN4是细胞质和细胞核中一种高度保守的肌动蛋白结合蛋白,具有多种调控作用,可激活各种转录因子(如NF-κB),NS7蛋白可能参与了这一过程[21-22]。
2 PDCoV的致病机制
2.1 PDCoV入侵细胞的分子机制
已有研究发现,病毒尖峰(S)糖蛋白的激活会触发细胞内的蛋白酶,确定了PDCoV进入宿主细胞的两条途径:细胞表面的胰蛋白酶和内体中的组蛋白酶L(CTSL)、组蛋白酶B(CTSB)。采用内吞体酸化抑制剂(Baf-A1)处理细胞可完全抑制PDCoV 进入;切断siRNA 介导的CTSL 或CTSB则可显著减少病毒感染,结果表明PDCoV可通过内吞途径进入细胞。
此外,使用胰蛋白酶处理细胞会导致S蛋白的裂解和激活,使PDCoV直接从细胞表面进入宿主细胞[8,23]。Yang等[24]研究发现,猪氨基肽酶N(pAPN)通过内吞途径促进PDCoV 入侵细胞并完成复制周期,阐明了pAPN 促进PDCoV入侵细胞及复制的分子机制。
2.2 PDCoV天然免疫应答
目前对PDCoV 致病机制的研究主要限于天然免疫。天然免疫是身体对入侵的病原微生物的第一道防线,主要由天然免疫分子介导,清除和消灭病原体。在众多天然免疫分子中,干扰素是一种具有广谱抗病毒活性的细胞因子。干扰素的抗病毒作用是通过与细胞表面的干扰素受体结合,激活细胞内信号通路(JAK-STAT),并通过一个放大的信号级联激活干扰素相关刺激基因(ISG)的表达。在长期适应宿主的进化过程中,病毒逐渐获得了中和天然宿主免疫的能力,并以免疫逃逸的形式更好地适应宿主细胞。当PDCoV 病毒介导细胞受体进入靶细胞后,其基因组RNA通过病毒-宿主膜融合释放到细胞质中(PDCoV间接影响宿主细胞抗病毒信号级联反应见图1)[25]。在细胞因子诱导阶段,内体TLR、胞质RIG-I 和MDA5 均可感觉到病毒来源的RNA基因组和其他复制的RNA中间体的存在。这些受体对病毒来源的RNA进行识别并激活一系列信号分子,导致转录因子NF-κB、IRF1、IRF3 和IRF7 的核转位。在细胞核内,这些转录因子与其各自的PRD区域结合,驱动Ⅰ型和Ⅲ型IFN 以及促炎细胞因子产生,并被分泌到细胞外。之后在信号转导阶段,Ⅰ型和Ⅲ型IFN以自分泌和旁分泌的方式与其同源受体结合,诱导JAK-STAT通路激活,导致ISGF3 复合体的核转位,并产生干扰素刺激基因(ISGs),加快了PDCoV的复制。
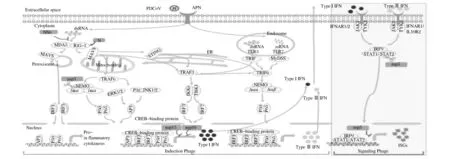
图1 PDCoV间接影响宿主细胞抗病毒信号级联反应Fig.1 PDCoV indirectly affects antiviral signaling cascade in host cells
有研究表明,PDCoV nsp5具有拮抗IFN-I信号转导的作用,并且该抑制作用依赖其蛋白酶活性[26]。进一步研究结果显示,PDCoV nsp5通过裂解STAT2抑制IFN-I 信号,同时感染该病毒内源性STAT2 也被裂解,所有STAT2 裂解的产物失去了诱导IFN-I 的能力。上述结果表明,PDCoV nsp5 对IFN-I 信号具有拮抗作用。此外,PDCoV的结构蛋白N蛋白可通过干预双链RNA 及蛋白激酶R活化蛋白与RIG-Ⅰ结合,以及抑制RIG-Ⅰ的K63 多聚泛素化,拮抗IFN-β 产生[27]。目前对PDCoV 致病机制的研究角度单一且数量较少,有待从多个角度进一步深入探索。
2.3 PDCoV诱导宿主细胞自噬、凋亡
细胞自噬是指细胞面临营养缺乏、应激、病原微生物入侵或缺乏生长因子时,可能在细胞质中形成自噬囊泡,包裹外来的病原微生物。之后损坏的细胞器和一些蛋白质被运送到溶酶体,形成自噬溶酶体,最终消化包裹的物质,并再生细胞器稳定细胞内环境。病毒和细胞自噬之间的联系引起了广泛关注,已证明体内许多免疫相关因素通过自噬抵抗病毒感染,但许多病毒也可利用细胞自噬加强其复制。此外,细胞凋亡是一个细胞主动终止其生命的过程,对维持细胞的平衡至关重要。细胞凋亡可由多种细胞信号触发,包括细胞内Ca2+浓度增加,氧化损伤导致的活性氧(如羟基自由基)积累,以及毒素、一氧化氮(NO)、生长因子和病毒感染等激素刺激,可在各种应激条件下发生,并在抵抗病毒感染中发挥重要作用。病毒感染宿主细胞是一个互动的过程,在此过程中,细胞通过细胞凋亡的过程消除病毒进而抵抗病毒感染,而病毒可延长细胞的存活时间,使病毒后代的数量最大化,或诱导细胞死亡,促进病毒后代的释放和传播,使其能够继续感染周围的细胞。
袁为峰[28]通过构建PDCoV 结构蛋白N、M、E 和非结构蛋白Nsp6 的真核表达质粒,证明了PDCoV 结构蛋白N和非结构蛋白Nsp6 均可显著上调自噬基因LC3-Ⅱ的表达。为进一步证实自噬和PDCoV 感染之间的关系,试验使用自噬促进剂雷帕霉素和自噬抑制剂氯喹预处理LLCPK1细胞和IPEC-J2细胞。结果表明,PDCoV N蛋白的表达水平随着LC3-Ⅱ蛋白的表达水平增加,而氯喹可通过自噬途径抑制PDCoV 复制,研究结果为进一步探索病毒与细胞自噬间的关系提供了更多理论依据。
PDCoV 通过粪便-口腔途径传播[28],并在小肠绒毛上皮的细胞质中复制,被感染的小肠肠道细胞发生广泛的空泡化并与绒毛上皮分离,导致绒毛萎缩。肠道细胞的大量损失阻碍了小肠对营养物质和电解质的吸收和消化,导致仔猪吸收不良和消化不良引起的腹泻,造成致命的脱水。这种病理生理变化(包括肠细胞的空泡变性和最终死亡)与病毒的细胞溶解作用引起的坏死有关,而非细胞凋亡。Jang 等[29]研究表明,PDCoV 只能在体外诱导ST 猪睾丸细胞和LLC猪肾细胞的凋亡,不能诱导体内感染的肠道细胞发生凋亡。Lee 等[30]阐述了PDCoV 感染后细胞凋亡过程的潜在机制,指出使用PAN-caspase抑制剂可抑制PDCoV诱导的细胞凋亡,减少PDCoV 的复制,提示PDCoV 诱导的细胞凋亡可能伴随着caspase 的激活。此外,PDCoV 感染需要激活caspase-9 启动子,该启动子负责内在的线粒体凋亡途径。试验结果表明,PDCoV感染导致Bax介导的线粒体外膜通透性(MOMP),导致线粒体细胞色素c(Cytc)特异性地重新定位到细胞质中。线粒体通透性转换孔(MPTP)开放的抑制剂环孢素A(CsA)可显著抑制PDCoV 引发的细胞凋亡和病毒复制。此外,在CsA 存在下,PDCoV 感染细胞中细胞色素c 的释放被完全抑制,提示MPTP 在PDCoV 感染的内源性细胞凋亡中起到关键作用。综上所述,PDCoV感染通过Bax募集或MPTP开放刺激MOMP,从而允许导致细胞凋亡的Cytc 释放到细胞质中,促进病毒的体外复制。
3 展望
近年来,新发冠状病毒如SADS-CoV、MERS 以及2019 年暴发的SARS-CoV-2 给人类和动物带来了严重的健康问题和经济损失。PDCoV 属于冠状病毒科中冠状病毒亚科的三角洲冠状病毒属,可感染鸟类和哺乳动物,是仅次于PEDV的猪肠道致病病毒,了解PDCoV编码蛋白的功能以及病毒如何调控宿主共能、利用宿主机制逃避免疫反应,有助于明确病毒的致病机制和商品化疫苗研发,减少养殖户的经济损失。
