哮喘中miRNA、lncRNA、circRNA、转录因子和靶基因的调控网络
2023-06-30李昱剑阚璇
李昱剑,阚璇
(天津医科大学总医院儿科,天津 300070)
支气管哮喘简称哮喘,是临床最常见的慢性气道炎症性疾病之一。哮喘复杂的发病机制和多样化的临床表现既增加了临床医生的诊治难度,也使患者在日常生活中饱受痛苦[1]。目前全球至少有3 亿哮喘患者,且哮喘的发病率在全球范围内仍呈上升趋势[2]。虽然随着医学进步,既往统计的哮喘死亡率已经有所下降,但从2006 年开始其下降趋势就出现了急刹车,表明现有治疗手段已经遇到了瓶颈[3]。
越来越多疾病的诊治从蛋白质水平进入到了转录和调控水平,其中非编码RNA 发挥着不可替代的作用[4]。研究表明,与健康人相比,哮喘患者外周血样本中lncRNA 的数量和表达存在差异[5]。与传统的线性RNA 不同,circRNA 呈现封闭的环状结构,使其表达更加稳定。circRNA 可以通过与哮喘等疾病相关的miRNA 相互作用,从而对哮喘等疾病的发生、发展起到重要作用[6]。转录因子是一组蛋白质分子,能使目标基因在特定的时间和空间上以特定的强度进行表达。一些研究也指出,转录因子可以参与哮喘的气道炎症、气道重塑和免疫调节[7]。
近年来,有关miRNA 与哮喘关系的研究层出不穷,对哮喘的诊治起到了积极的推动作用,但关于lncRNA、circRNA、转录因子等非编码RNA 与哮喘关系的研究仍有较多空白。本研究将使用上述方法筛选哮喘的DEGs,并构建miRNA、lncRNA、circRNA、转录因子和靶向药物与哮喘靶基因之间的调控网络。对哮喘相关的遗传物质、功能、通路和靶向药物的探索,可以为哮喘生物标志物挖掘和精准医疗提供参考。
1 材料与方法
1.1 微阵列数据 本研究使用检索式:(′asthma′[MeSH Terms] OR ′asthma′[All Fields])AND(′Homo sapiens′[Organism]AND′Expression profiling by array′[Filter])从GEO[8](https://www.ncbi.nlm.nih.gov/geo/)数据库中筛选出了GSE43696 和GSE64913 这两个数据集,并从中提取了相应的临床信息。GSE43696来源于GPL6480 平台(Agilent-014850 Whole Human Genome Microarray 4x44K G4112F),包含88 例哮喘患者的支气管上皮细胞样本和20 名健康人体样本[9]。GSE64913 来源于GPL570 平台([HG-U133_P lus_2] Affymetrix Human Genome U133 Plus 2.0 Array),包含15 例哮喘患者的支气管上皮细胞样本和19 名健康人体样本[10]。
1.2 识别差异表达基因 通过核对两个数据集的信息来平均或删除没有相应基因符号的探针组和有多个探针组的基因。使用R 软件的Limma 包(版本:3.40.2)去除批次效应并识别DEGs。以“P<0.05 and/Fold Change/>1.5”作为筛选DEGs 的阈值。
1.3 GO 和KEGG 富集分析 R 软件中的Cluster-Profiler 包(版本:4.0)被用于GO 和KEGG 富集分析。箱式图由ggplot2 包绘制;PCA 图由ggord 包绘制;热图由pheatmap 包绘制。上述所有分析方法和R 软件包均由R 软件(2020)4.0.3 版完成。
1.4 Metascape 使用Metascape[11](http://metascape.org/)对得到的DEGs 再次进行GO 和KEGG 富集分析,与ClusterProfiler 包分析得到的结果进行对比验证和互相补充,使最终结果更加真实可靠。筛选条件:Min Enrichment=1.5,P<0.01 and Min overlap=3。
1.5 WEB-based GEneSeTAnaLysis Toolkit(WebGestalt)数据库和Reactome Pathway 数据库 使用WebGestalt[12](http://www.webgestalt.org/)和Reactome[13](https://reactome.org/)数据库对DEGs 从另一种算法角度进行GO 和KEGG 富集分析,从而补充单一算法的不足。WebGestalt 数据库的筛选标准是Number of IDs in the category:5-2000,FDR Method:BH,and Significance Level:FDR<0.05。Analysis Tools是Reactome 数据库中的一个分析工具,被用于通路研究。
1.6 蛋白-蛋白交互网络构建与靶基因筛选 使用STRING[14](https://string-db.org/)数据库构建DEGs的蛋白-蛋白交互网络,minimum required interaction score=0.4 被认为具有统计学意义。使用Cytoscape 对蛋白-蛋白交互网络进行可视化[15],并使用Cytoscape 的插件Cytohubba 筛选出最重要的9个核心基因。
1.7 lncRNA-miRNA-靶基因相互作用网络分析在Tarbase[16](http://carolina. imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex)和TargetScan[17](http://www.targetscan.org/vert_80/)数据库中,使用评分最高的4 个核心基因预测可能的miRNA,并通过取交集的方法降低结果的偶然性,从而得到可靠性最高的miRNA。使用ENCORI[18](https://starbase.sysu.edu.cn/)预测与miRNA 相匹配的lncRNA,并使用LncBase[19](http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-experimental)进行验证,从而构建出最可靠的lncRNA-miRNA-靶基因相互作用网络。
1.8 circRNA-miRNA-靶基因相互作用网络分析 使用与1.7 相同的方法预测筛选可以与9 个靶基因相匹配的miRNA。使用ENCORI 数据库预测与miRNA 匹配的circRNA,并使用circad 数据库[20](https://clingen.igib.res.in/circad/)进行临床验证。
1.9 转录因子-miRNA-靶基因相互作用网络分析 使用AnimalTFDB[21](http://bioinfo.life.hust.edu.cn/AnimalTFDB/)预测得分最高的4 个基因所对应的转录因子,并使用JASPAR[22](https://jaspar.genereg.net/)对得到的转录因子进行二次验证,以提高结果的可靠性。最终,每个靶基因筛选出了2 个评分最高的转录因子,筛选标准:Strand:+,P-value<0.05 and Q-value <0.05。
1.10 The Drug Gene Interaction(DGIdb)数据库 DGIdb[23](http s://www.dgidb.org/)被用于预测9 个靶基因的潜在靶向药物。筛选标准:Source Databases=22,Gene Categories=43,Interaction Types=31。
1.11 临床意义验证 提取GSE41649 数据集中的临床信息后,分别借助pROC 包和ggplot2 分析评分最高的4 个核心基因对哮喘的疾病预测能力和差异表达情况。
2 结果
2.1 哮喘差异表达基因的识别 使用R 软件的Limma 包(版本:3.40.2)对GSE43696 和GSE64913数据集的信息进行标准化和去批次效应后,共得到76 个DEGs,其中44 个基因上调,32 个基因下调(P<0.05 &/Fold Change/>1.5)。数据标准化后的箱式图、去批次前/后的PCA 图、DEGs 的火山图、热图及DEGs 中的特定基因如图1 所示。
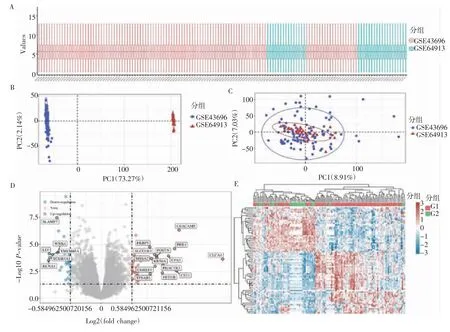
图1 哮喘差异表达基因的识别Fig 1 Identification of differentially expressed genes in asthma
2.2 GO 和KEGG 富集分析 使用R 软件的ClusterProfiler 包(版本:4.0)以及Metascape、WebGestalt 和Reactome 对DEGs 进行GO 和KEGG 富集分析并可视化(图2、3)。GO 富集分析显示,DEGs在淋巴细胞趋化、器官或组织特异性免疫反应、有机羟基化合物运输、细胞杀伤、黏膜免疫反应、抗菌体液反应、内吞调节等方面明显富集。KEGG 富集分析表明,DEGs 主要参与白细胞介素-17 信号通路、抑制一氧化氮产生、激活C3 和C5、果糖和甘露糖代谢等方面。

图2 上调和下调的差异表达基因的GO 和KEGG 富集分析Fig 2 Gene Ontology(GO)and Kyoto Encyclopedia of Genes and Genomes(KEGG)enrichment analysis of up-regulated genes and downregulated genes
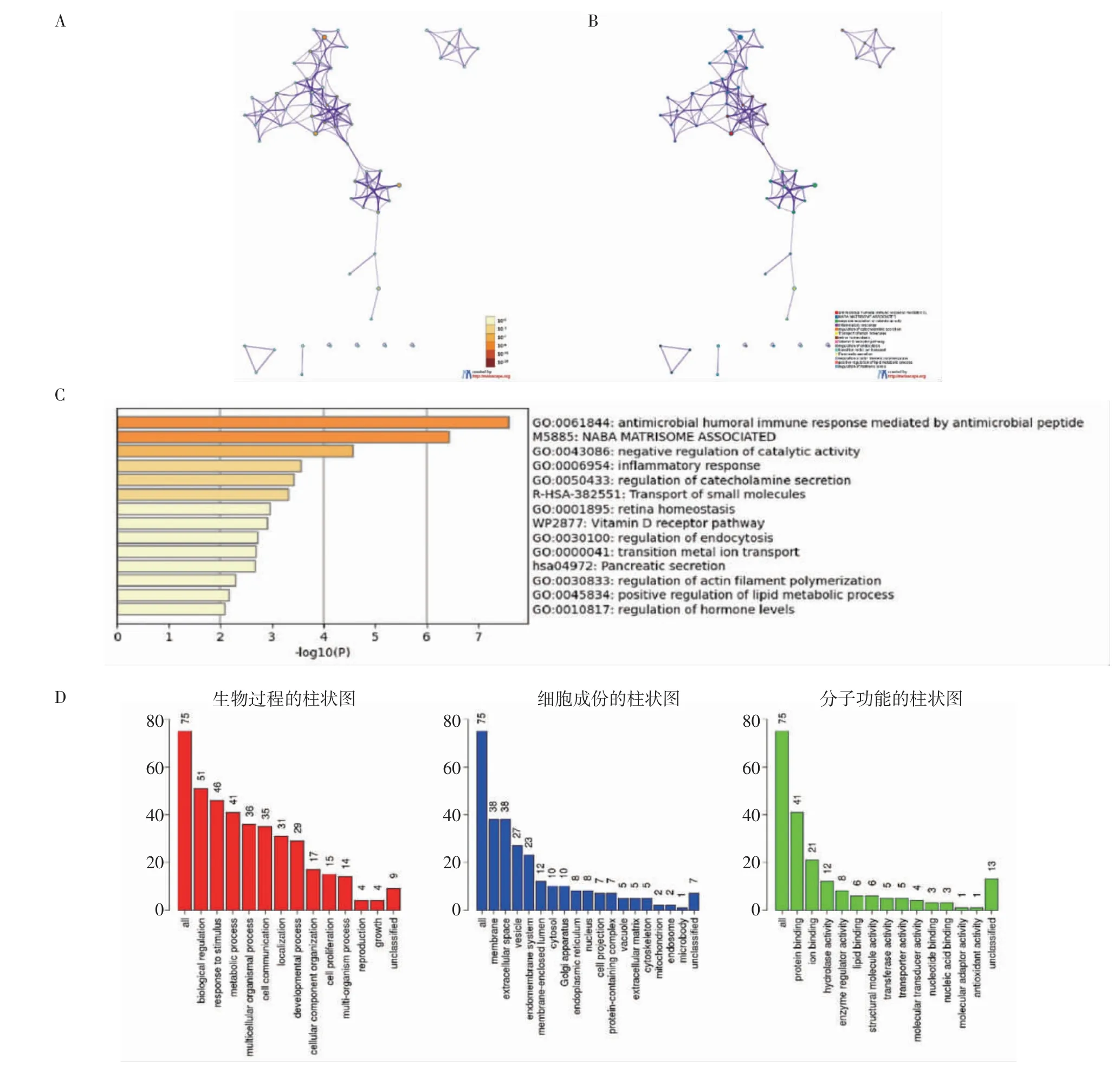
图3 差异表达基因的GO 和KEGG 富集分析Fig 3 Gene Ontology(GO)and Kyoto Encyclopedia of Genes and Genomes(KEGG)enrichment analysis of differentially expressed genes
2.3 蛋白-蛋白交互网络构建与靶基因筛选 使用Cytoscape 对STRING 构建的蛋白-蛋白交互网络进行可视化,共有43 个节点和54 条边(图4A)。通过Cytohubba 使用11 种方法来识别DEGs 中的核心基因,MCC 展现出了更好的比较性能。最终得到了9个评分最高的DEGs,它们分别是CLCA1、POSTN、CPA3、LTF、PIP、FKBP5、CCL26、SERPINB2 和 KIT(图4B)。在这9 个核心基因中,LTF 和PIP 在哮喘患者中低表达,其余基因均为高表达。
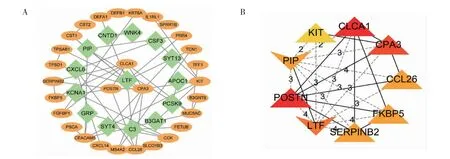
图4 蛋白-蛋白交互网络Fig 4 Protein-protein interaction network
2.4 lncRNA-miRNA-靶基因相互作用网络分析 评分最高的4 个核心基因分别是CLCA1、POSTN、CPA3 和LTF。Tarbase 和TargetScan 数据库预测到了67 个miRNA,其中hsa-miR-19b-3p 是两个数据库取交集后的公共结果。使用ENCORI 预测可能与hsa-miR-19b-3p 相互作用的lncRNA,并通过LncBase 验证结果。最终共得到7 个最可靠的lncRNA(ENSG00000272264、ENSG00000270087、ENSG00000245532、ENSG00000275764、ENSG0000-0263753、ENSG00000229807 和ENSG00000230590),并通过Cytoscape 对结果进行了可视化(图5A)。

图5 lncRNA/circRNA-miRNA-靶基因的相互作用网络Fig 5 The lncRNA/circRNA-miRNA-mRNA interaction network
2.5 circRNA-miRNA-靶基因相互作用网络分析 共有158 个miRNA 被预测到,最终筛选保留了2 个最可靠的miRNA(hsa-miR-19b-3p 和hsa-miR-218-5p)。使用ENCORI 预测可能与以上2 个miRNA相互作用的circRNA,共得到1 314 个circRNA。借助Circad 数据库验证预测得到的circRNA 的临床信息,最终确认SNX13 是与哮喘相关的circRNA(图5B)。
2.6 转录因子-miRNA-靶基因相互作用网络分析 用AnimalTFDB 预测评分最高的4 个核心基因所对应的转录因子,用JASPAR 对结果进行验证。最终,每个核心基因筛选出了2 个最可靠的转录因子。它们分别是SPI1、RREB1、AR、BCL6、IRF5、ZNF143、MAZ 和PAX5。转录因子-miRNA-靶基因相互作用网络见图6A。
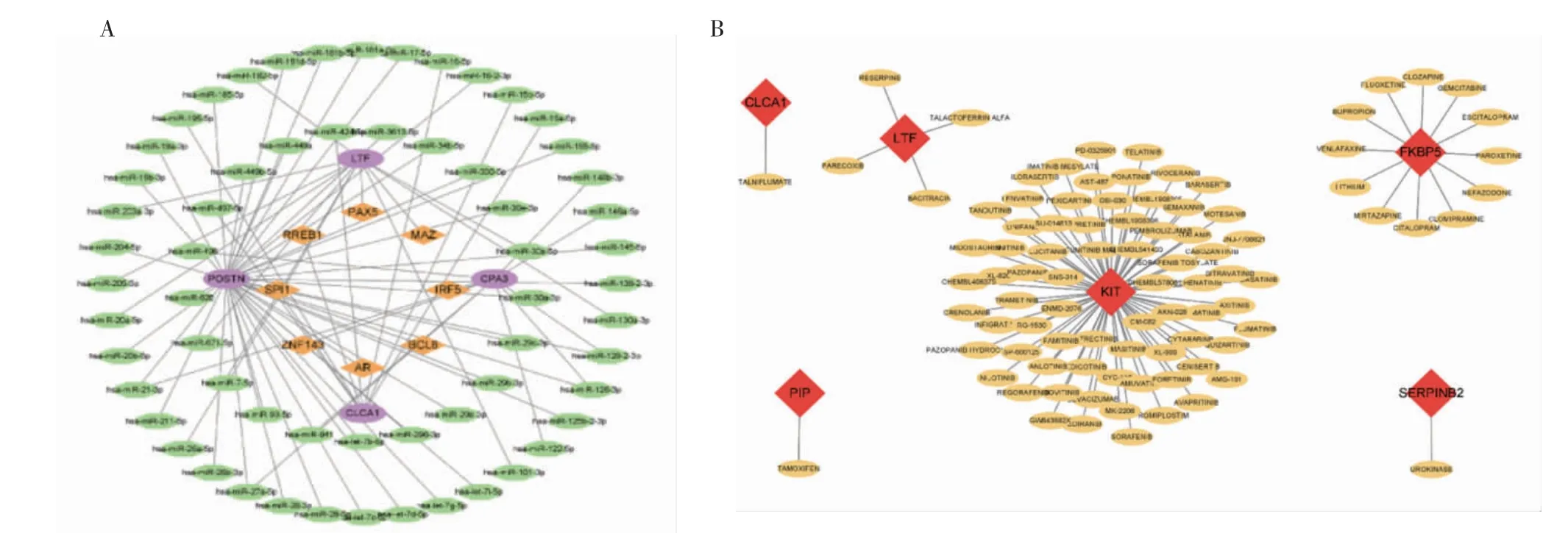
图6 转录因子-靶基因-miRNA 的相互作用网络与药物-靶基因的相互作用网络Fig 6 Transcription factor-mRNA-miRNA interaction network and drug-mRNA interaction network
2.7 哮喘的靶向药物 使用DGIdb 预测9 个靶基因的潜在靶向药物,共得到了91 种药物,这些药物有可能干预哮喘DEGs 的表达(图6B)。评分最高的4 个核心基因中有2 个预测到了潜在的靶向药物或分子化合物。CLCA1 的靶向药物是他尼氟酯,LTF的潜在靶向药物或分子化合物分别是α-苯丙氨酸转铁蛋白、帕瑞昔布、巴比妥珠和雷瑟平。此外,相互作用评分最高的药物——尿激酶被发现与SERPINB2 存在相互作用。预测到靶向药物或分子化合物最多的靶基因是KIT,它共有72 种靶向药物或分子化合物。
2.8 临床意义验证 ROC 曲线显示,评分最高的4 个hub 基因中,CLCA1、POSTN 和LTF 对哮喘均具有较高的疾病预测能力,而CPA3 同样具有中等的疾病预测能力(图7A~7D)。分组比较图显示CLCA1、POSTN 和LTF 在新的临床数据中同样存在表达差异,其中CLCA1 和POSTN 在哮喘中高表达,LTF 在哮喘中低表达(图7E~7H)。

图7 核心基因的ROC 曲线和分组比较图Fig 7 ROC curves and grouping comparison plots for hub genes
3 讨论
哮喘的发病机制复杂,在诊断和精准医疗方面仍存在诸多不足。传统治疗药物如糖皮质激素等的治疗周期长、依从性差,治疗效果并不足够理想,而奥马珠单克隆抗体等生物制剂的安全性和经济负担让很多患者及家属感到担忧[24]。本研究构建了miRNA、lncRNA、circRNA、转录因子、靶向药物和靶基因的调控网络,从而为哮喘相关生物标志物的探索提供一定的参考依据,对哮喘患者和有潜在风险者的早期识别、早期诊断和早期治疗有一定的临床价值。
本研究通过对GSE43696 和GSE64913 这2 个数据集的分析,得到了44 个在哮喘中高表达的DEGs 和32 个低表达的DEGs。GO 分析显示DEGs 的功能主要富集在先天免疫、获得性免疫和炎症的发生、发展上,尤其是细胞杀伤、内吞作用的调节和淋巴细胞趋化作用等方面。既往研究表明,一些哮喘相关基因可以促进气道炎症,并在炎症体的帮助下诱发哮喘的发生。此外,既往感染引起过敏原特异性免疫功能的过早表达,对哮喘的发生、发展也有一定作用[25],这与本研究的结论一致。通路富集的结果显示,趋化因子信号通路、激素和有机物代谢通路和免疫调节通路在DEGs 中具有较好的富集结果,说明哮喘的发生发展与免疫、微生物、炎症以及有机物代谢密切相关,提示可以从这些角度对哮喘的诊断和治疗进行早期干预。
在构建DEGs 的蛋白-蛋白交互网络后,共筛选出了9 个靶基因,其中LTF 和PIP 在哮喘患者中低表达,其余为高表达。4 个核心基因,即CLCA1、POSTN、CPA3 和LTF 的筛选评分最高。CLCA1 的全称是氯通道附属物1,可以调节钙激活的氯离子传导,产生分泌蛋白和膜相关蛋白,这些蛋白可以增加白细胞浸润和气道高反应性,从而增加哮喘的易感性[26]。POSTN 是骨膜蛋白,它在转化生长因子-β 和白细胞介素-13 的作用下产生。据报道,在哮喘患者的呼气冷凝物中可以检测到骨膜蛋白,且POSTN 高表达的患者肺功能相应下降,表明POSTN 有被用作生物标志物的可能性[27]。CPA3 的全称是羧肽酶A3,有报道称该基因与哮喘、冠心病、COVID-19 等有关,原因可能是该酶可参与巨噬细胞的激活和促炎症细胞因子的上调,从而直接或间接参与炎症和免疫调节[28]。LTF(乳铁蛋白)是转铁蛋白家族基因的一员,其蛋白产物是免疫系统的重要组成部分,然而遗憾的是目前还没有发现这个基因与哮喘发生和发展之间的具体关系。本研究筛选得到的这些基因具有作为哮喘诊治靶基因的巨大潜力。
Has-miR-19b-3p 和hsa-miR-218-5p 是通过不同的数据库和算法最终筛选得到的2 个miRNA。既往研究显示,hsa-miR-19b-3p 可能在哮喘的发生和发展中发挥潜在作用[29]。Hsa-miR-218-5p 是由KIT 预测得到的,同样有文献表明它可能在嗜酸性粒细胞气道炎症中起到保护作用[30]。在miRNA 的基础上,共筛选得到了7 个lncRNA 和1 个circRNA,最终构建了miRNA、lncRNA、circRNA 和靶基因的表达调控网络,为哮喘诊断的进一步研究和精准医疗提供了依据。
共筛选得到了8 个转录因子,它们在炎症、细胞增殖和分化、免疫调节、生长发育等方面发挥着重要作用[31-32],有被用作哮喘治疗标志物的可能性。此外,靶向药物的预测显示CLCA1 的靶向药物他尼氟酯是一种非甾体类抗炎药,可用于纤维囊肿和哮喘的辅助治疗[33]。LTF 的潜在靶向药物或分子化合物分别是α-苯丙氨酸转铁蛋白、帕瑞昔布、巴比妥珠和雷瑟平,它们在治疗哮喘方面的作用尚不清楚。交互得分最高的靶向药物尿激酶主要作用于内源性纤维蛋白溶解系统,它也可能与哮喘的治疗有关[34]。此外,靶基因KIT 共预测到了72 种靶向药物,这些药物同样可能为哮喘的治疗提供一个新的方向,所以值得进一步研究和探索。
本研究使用了2 个去批次效应后的数据集来增加样本量,并使用了1 个新的数据集验证最终的结果,从而使本文结论更加可靠。此外,本研究还预测了与哮喘有关的转录因子和药物,这使本研究比以往的研究更加全面和广泛。本研究同样存在一些局限性。首先,虽然使用了多个数据库和不同算法取交集的方法来提高结果的可靠性,但仍缺少体内体外实验对通路和机制进行进一步验证,后续可以从实验验证等方面对本文的研究结果进行更加深入的分析研究。此外,本研究的数据来自于公共数据库,缺少外部数据进行验证,因此存在假阳性的可能性,后续可以使用更高质量的外部数据进行前瞻性研究与本文结果进行互相佐证从而降低假阳性率。
综上所述,本研究共识别出了76 个DEGs,并从中筛选出了9 个靶基因。细胞杀伤、调节内吞作用、淋巴细胞趋化等生物功能和趋化因子信号通路、免疫调节通路等通路都在哮喘的发生、发展中起到一定的作用。SNX13 和7 个lncRNA 通过hsa-miR-19b-3p 和hsa-miR-218-5p 参与哮喘相关基因的表达和调控。此外SPI1 等转录因子和他尼氟酯等药物同样可能会干预哮喘相关基因的表达调控。
