阿莫西林颗粒中有关物质高效液相色谱分析方法的建立
2023-06-20郑如程田胜程正江跃张德兵
郑如程,田胜,程正,江跃,张德兵
阿莫西林(Amoxicillin)称羟氨苄青霉素,其为白色或类白色的结晶性粉末,稍有特异的气味和苦味,是第二代青霉素的主要品种,系广谱半合成抗生素,具有广谱抗菌作用,世界卫生组织(WHO)推荐本品作为首选的β-内酰胺类口服抗生素,在口服抗生素中占有重要的位置[1]。本品临床上应用于革兰阴性菌、革兰阳性菌等所致的感染性疾病、呼吸系统疾病及泌尿系统疾病[2-5]。由于其高效、低毒,因此在临床上应用十分广泛,但其不良反应发生率居各类药物之首。为了减少不良反应的发生[6-7],必须对其杂质进行控制。阿莫西林原料[8-9]、阿莫西林注射剂[10-11]、阿莫西林胶囊[12-14]中的杂质研究已有不少文献报道,但对阿莫西林颗粒[15-16]中杂质的研究相对较少。
经检索各国药典,现行《中华人民共和国药典》(ChP)2015 年版和2020 年版中“阿莫西林颗粒”标准中未收录“有关物质”检查项[17-18],国外药典中也未收载有阿莫西林颗粒剂,其相似剂型为“阿莫西林口服混悬剂”,但均未控制有关物质,其中美国药典USP41“阿莫西林口服混悬剂”征求意见稿[19]中控制了“有关物质”检查项,但正式稿及美国药典USP42[20]中均未控制。
本研究于2021 年6 月至2022 年2 月,结合原料药生产工艺,检索各国药典,阿莫西林目前已知的杂质共有17 个(见表1)。其中杂质H 与杂质P 这两种杂质在原料合成工艺中不产生,阿莫西林颗粒制剂中将不再研究;杂质N为工艺杂质和副产物,是6-APA与杂质D的缩合物,杂质O为降解产物,是杂质D 与阿莫西林的三聚合物,两者在聚合物测定中可予以控制,故按照其他单个杂质控制;对可以获得对照品的杂质A、B、C、D、E、F、G、I、J、K、L、M、DHPM这13种杂质在阿莫西林颗粒中进行了研究,对实验条件进行了优化,使阿莫西林颗粒中主成分与各杂质可实现良好分离;并进行相关的方法学验证。
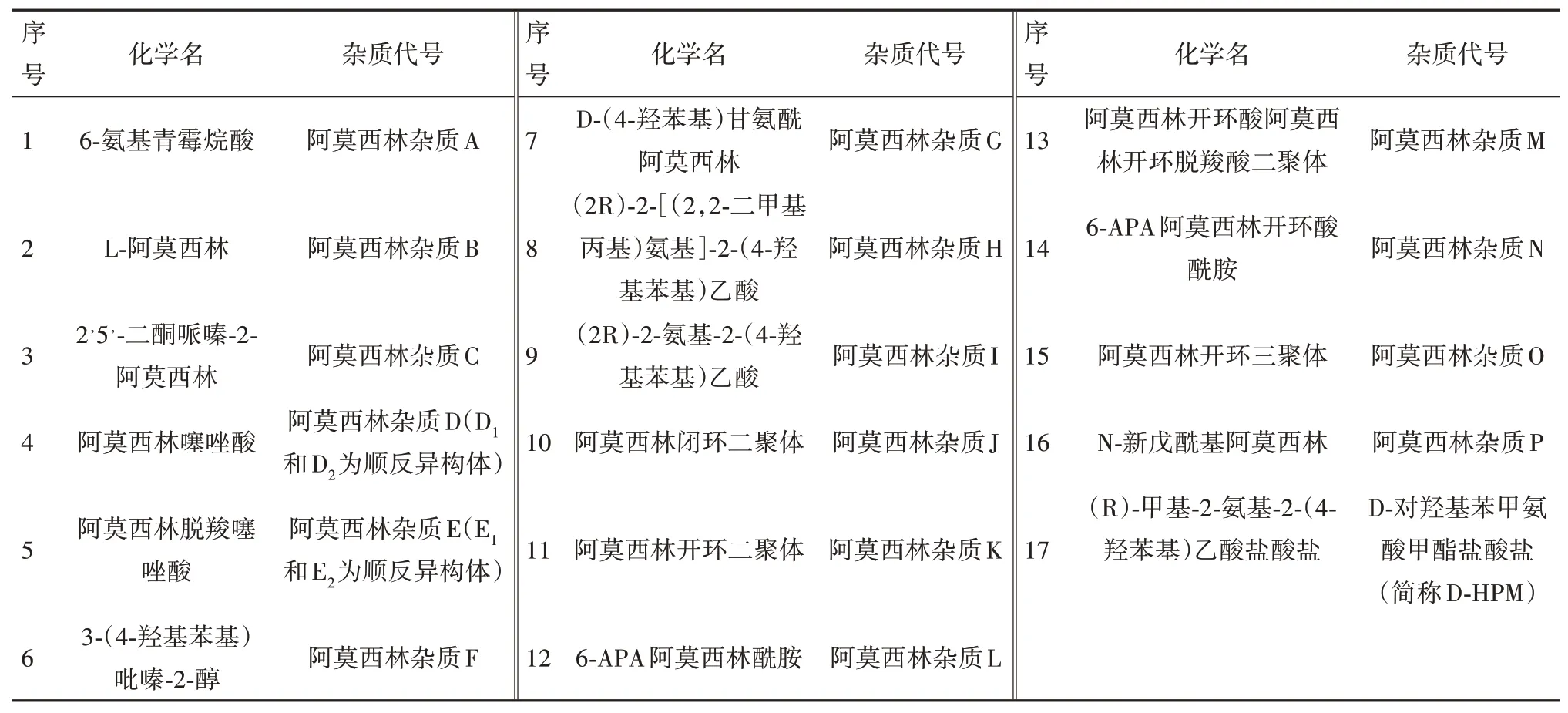
表1 阿莫西林各杂质汇总一览表
1 仪器与试药
SQP QUINTIX 224-1CN 电子天平(德国赛多利斯集团),S210 型pH 计(瑞士梅特勒-托利多集团),LC-20A 型高效液相色谱仪(二极管阵列检测器,日本岛津公司)。
阿莫西林颗粒(A公司:批号S200301,S200302,S 200303;L 公司:批号1903A0,9420),阿莫西林对照品(中国食品药品检定研究院,批号130409-201512,含量86.9%),阿莫西林工作对照品(A 制药公司,批号190121034,含量87.1%),阿莫西林系统适用性对照品(中国食品药品检定研究院,批号130608-201804),杂质A(中国食品药品检定研究院,批号130595-201001,含量100%),杂质B(USP对照品,批号R081J0,含量100%),杂质C(USP 对照品,批号R072H0,含量100%),杂质D(USP 对照品,批号R051S0,含量100%),杂质E(美国CATO 公司,批号C4X-11486-1708,含量95.6%),杂质F(USP 对照品,批号R074A0,含量100%),杂质G(USP 对照品,批号R019S0,含量100%),杂质I(中国食品药品检定研究院,批号130435-201903,含量99.9%),杂质J(美国CATO 公司,批号161-12-6,含量90.1%),杂质K(美国CATO 公司,批号C4X-114816-1812,含量97.7%),杂质L(加拿大TRC 公司,批号2-JPD-178-1,含量95%),杂质M(加拿大TRC 公司,批号4-JPD-26-1,含量90.0%),D-HPM(美国CATO 公司,批号166-12-23,含量98.7%)。
乙腈为色谱纯,其余试剂均为分析纯,试验用水由Milli-Q系统制备。
2 方法与结果
2.1 色谱条件色谱柱:Agilent Zorbax SB-C8 柱(4.6 mm×250 mm,5 µm);鬼峰捕集小柱:Welch Ghost-Buster Column(4.6 mm×50 mm,5 µm);流动相A 为6.8 g/L 磷酸二氢钾溶液(用20% 氢氧化钠溶液调节pH 至5.5±0.1);流动相B 为6.8 g/L 磷酸二氢钾溶液(用20 %氢氧化钠溶液调节pH 至4.5±0.05)-乙腈(84∶16);按表2 进行梯度洗脱程序;检测波长230 nm;柱温30 ℃;流速2.0 mL/min;低温进样器温度4 ℃;进样量20 µL;溶剂为流动相A。

表2 梯度洗脱程序
2.2 溶液配制供试品溶液:取A 公司阿莫西林颗粒适量,精密称定,冷水中用流动相A溶解并定量稀释制成每1 毫升中约含阿莫西林1.5 mg 的溶液,滤过,取续滤液即得。
对照溶液:精密量取供试品溶液1 mL,置100mL量瓶中,用流动相A稀释至刻度,摇匀,即得。
空白辅料溶液:按处方比例取空白辅料,同供试品溶液配制方法制备,即得。
阿莫西林对照品贮备溶液:精密称取阿莫西林工作对照品,加溶剂溶解并定量稀释制成浓度约为0.5 g/L的贮备溶液。
杂质贮备溶液:精密称取各有关杂质对照品,加溶剂溶解并定量稀释制成各杂质对照品浓度约为0.3 g/L的杂质贮备溶液。
杂质单标溶液:各精密量取上述各杂质贮备溶液(杂质D、E、J 贮备溶液为1.0 mL,其余均为0.5 mL),分别用溶剂定容至20 mL,作为杂质单标溶液。
阿莫西林+杂质混合对照溶液:取阿莫西林颗粒约240 mg,精密称定,置20 mL 量瓶中,加溶剂适量,冷水超声15 min,精密加入各杂质贮备溶液(加入杂质D、E、J 贮备溶液为1.0 mL,其余均为0.5 mL),用溶剂定容,摇匀,滤过,取续滤液,即得。
2.3 系统适用性试验取阿莫西林系统适用性对照品适量,加流动相A溶解并稀释制成每l毫升中约含1.5 mg 的溶液,作为系统适用性溶液1,注入液相色谱仪,记录色谱图,见图1。

图1 阿莫西林系统适用性溶液1色谱图
取阿莫西林对照品与杂质E 对照品适量,加流动相A 溶解并稀释制成每1 毫升中约含阿莫西林1.5 mg、杂质E约0.03 mg的混合溶液,作为系统适用性溶液2,注入液相色谱仪,记录色谱图,见图2。由试验结果可知,阿莫西林主峰与各有关物质峰分离度良好,且杂质峰不干扰主峰与各有关物质峰定位。

图2 阿莫西林系统适用性溶液2色谱图
2.4 专属性试验
2.4.1辅料干扰试验 取“2.2”项下供试品溶液和空白辅料溶液分别注入液相色谱仪,记录色谱图,见图3,4。

图3 阿莫西林供试品溶液图谱

图4 阿莫西林颗粒空白辅料溶液图谱
由试验结果可知:主峰与相邻杂质及各杂质间均达到基线分离,各杂质分离度符合要求;空白辅料出峰,但不干扰本品有关物质的测定。
2.4.2杂质定位 取“2.2”项下各杂质单标溶液和阿莫西林+杂质混合对照溶液,分别注入液相色谱仪,色谱图结果显示,各杂质间分离良好且峰行较好。结果详见表3,图5。

图5 阿莫西林+杂质混合对照溶液色谱图

表3 杂质定位专属性试验结果
2.4.3强制降解试验 取供试品及供试品溶液,分别进行酸、碱、氧化、高温、光照、紫外破坏(强制降解条件见表4),取上述各破坏溶液及未破坏的样品溶液,除梯度洗脱时间延长为保证杂质峰出峰完全外,其余均按照“2.1”项下色谱条件,分别注入高效液相色谱仪,记录色谱图,见图6~10。由试验结果及色谱图可知:酸破坏的主要降解杂质为杂质D2;碱破坏的主要降解杂质为杂质D2与杂质C;氧化破坏的主要降解杂质为杂质D2与杂质B;高温液体破坏的主要降解杂质为杂质C;紫外液体破坏的主要降解杂质为杂质D1与杂质C;其余降解试验样品状态比较稳定,未降解出较大杂质;各个强制降解条件下产生的杂质与主峰均能有效分离;各强制降解试验中所得色谱图中主峰纯度均为1.00,符合验证要求。

图6 阿莫西林供试品溶液酸破坏试验图谱
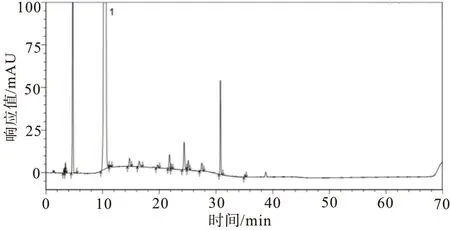
图7 阿莫西林供试品溶液碱破坏试验图谱

图8 阿莫西林供试品溶液氧化破坏试验图谱

图9 阿莫西林供试品溶液高温液体破坏试验图谱
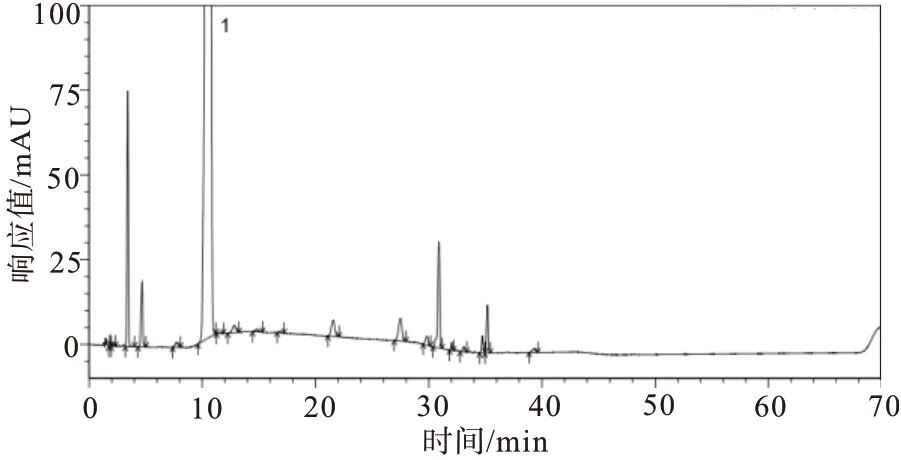
图10 阿莫西林供试品溶液紫外液体破坏试验图谱

表4 阿莫西林供试品溶液强制降解试验条件
2.5 检测限(LOD)、定量限(LOQ)、线性关系和校正因子测定取“2.2”项下各杂质贮备溶液及阿莫西林对照品贮备溶液,逐步稀释,配制成各杂质单标溶液浓度20%、60%、100%、140%、200%的线性溶液,注入高效液相色谱仪,记录色谱图,以峰面积(A)对浓度(C:µg/mL)进行线性回归,计算LOD、LOQ和校正因子,结果见表5。
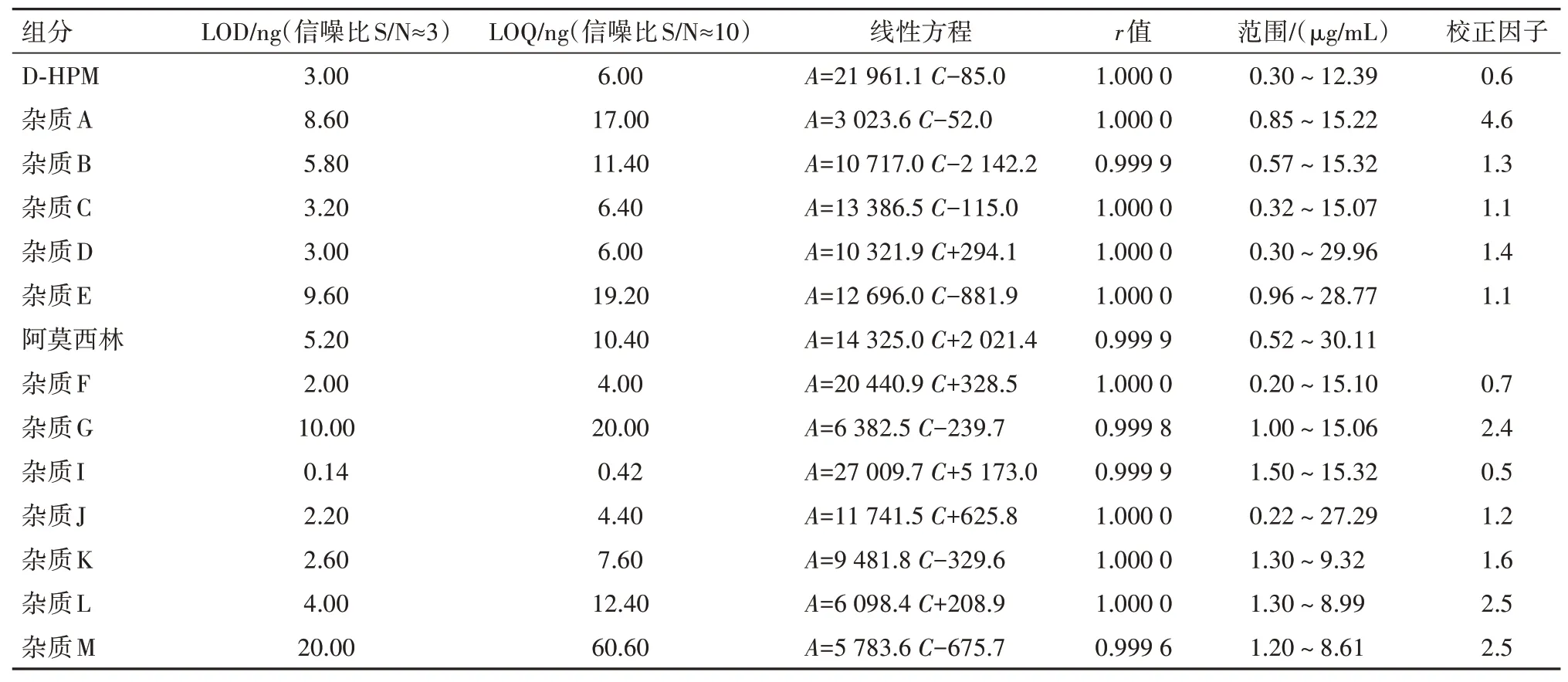
表5 阿莫西林及各杂质的标准曲线、LOD、LOQ与校正因子
2.6 精密度测定取“2.5”项下100%浓度的线性溶液,连续进样6 针,计算主峰面积和保留时间的RSD 值。阿莫西林和各杂质的峰面积RSD 值均小于2.0 %,保留时间的RSD 值均小于1.0%,表明方法的精密度良好。
2.7 稳定性试验取“2.2”项下各杂质贮备标溶液及阿莫西林对照品贮备溶液,配制成对照品稳定性试验溶液,于低温(4 ℃)放置12 h,分别在0 h、2 h、4 h、6 h、8 h、10 h 和12 h 时,取样检测,考察峰面积的RSD 值。结果显示,在12 h 内,阿莫西林主峰峰面积RSD 为0.5%,杂质A 和杂质C 峰面积RSD 最小,均为0.4%,杂质B 峰面积RSD 最大,为2.0%。各杂质低温4 ℃放置12 h 下,峰面积的RSD 均未超过2.0%,说明于低温4 ℃条件下放置12 h 内稳定。
取“2.2”项下空白辅料溶液和供试品溶液,于低温(4 ℃)放置8 h,分别在0 h、1 h、2 h、3 h、4 h、6 h和8 h 时,取样检测。结果显示,供试品溶液于低温4 ℃下放置4 h,杂质D 变化量为0.024%,杂质J变化量为0.004%,其他最大单个杂质的变化量为0.006%,总杂质的变化量为0.022%,说明供试品溶液于低温4 ℃下放置4 h内稳定。
2.8 重复性和中间精密度试验取同一批阿莫西林颗粒样品,按“2.2”项下方法制备供试品溶液6份,取样检测,计算各杂质的变化量。结果显示,6次测定结果中,杂质D 变化量为0.050%,杂质J变化量为0.011%,其余各杂质的变化量小于0.05%,总杂质变化量为0.056%,小于0.1%。
2.9 回收率试验取同一批阿莫西林颗粒,按“2.2”项下方法制备供试品溶液,分别加入各杂质单标溶液浓度50%、80%、100%、120%的杂质贮备溶液,每个含量平行配制三份,分别注入液相色谱仪,记录色谱图,计算回收率。各浓度下,杂质D平均回收率均在90%~110%,杂质J和杂质E平均回收率均在92%~105%,其余各杂质平均回收率均在90%~108%。试验结果见表6。
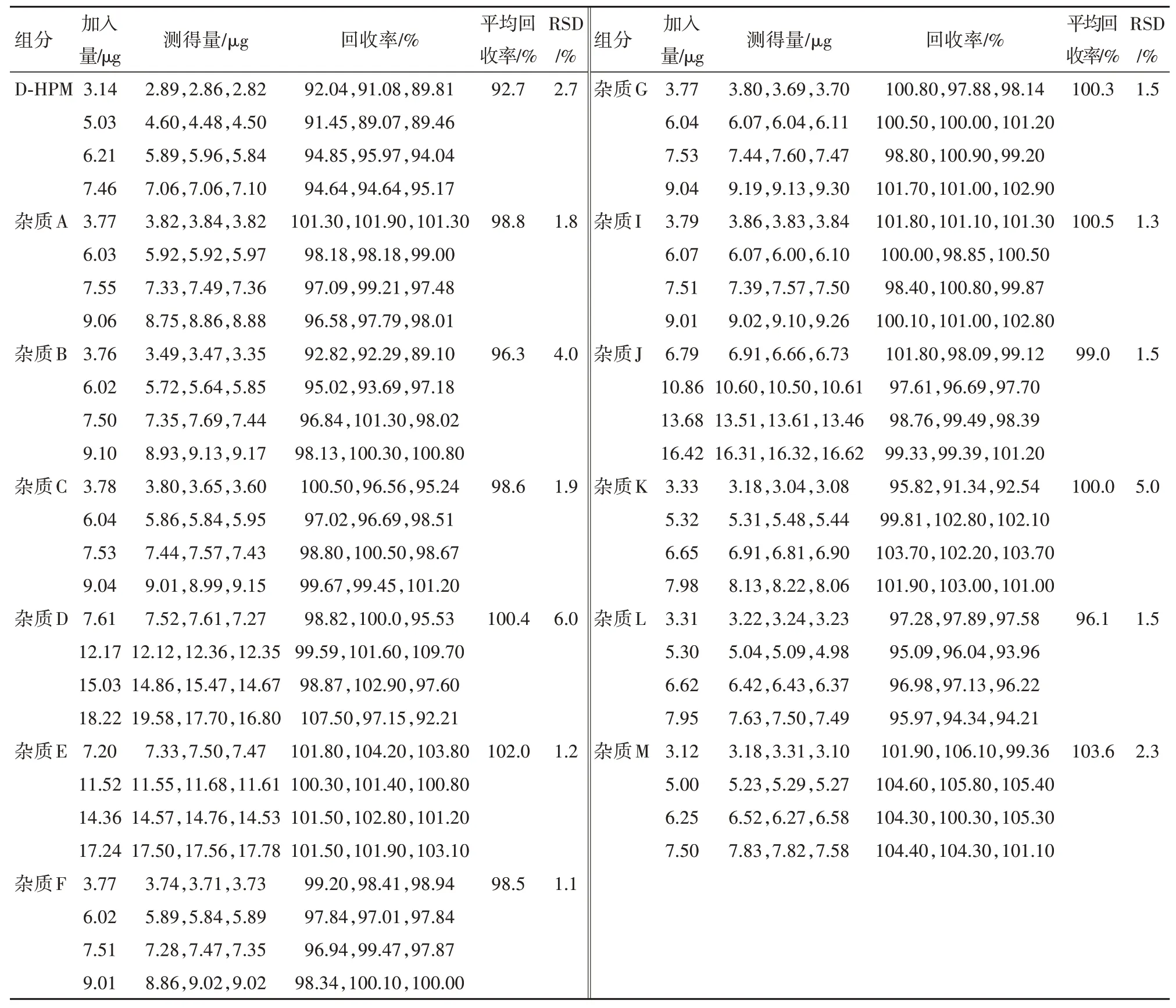
表6 阿莫西林供试品回收率试验结果
2.10 耐用性试验在其他条件不变的情况下,分别微调检测波长(±5 nm)、盐溶液pH(流动相A:pH±0.2,流动相B:pH(±0.05)、柱温(±5 ℃)、流速(±0.2 mL/min)及更换同型号同规格不同品牌色谱柱,测定本品有关物质,以分离度和各杂质的变化量考察方法的耐用性。在各耐用性色谱条件下,杂质混合溶液中各杂质均能达到基线分离且主峰与相邻峰的分离度均符合要求;供试品中单个杂质变化量均小于0.1 %;总杂质变化量均小于0.2 %;说明本法耐用性良好。
2.11 样品测定取3 批次A 公司生产的阿莫西林颗粒,同时取2 批次L 公司生产的阿莫西林颗粒作为参比制剂,按“2.1”项下色谱条件进样分析,记录色谱图,分别按自身对照法和外标法计算杂质含量,试验结果见表7,8。

表7 阿莫西林样品测定结果(主成分自身对照加校正因子法)/%
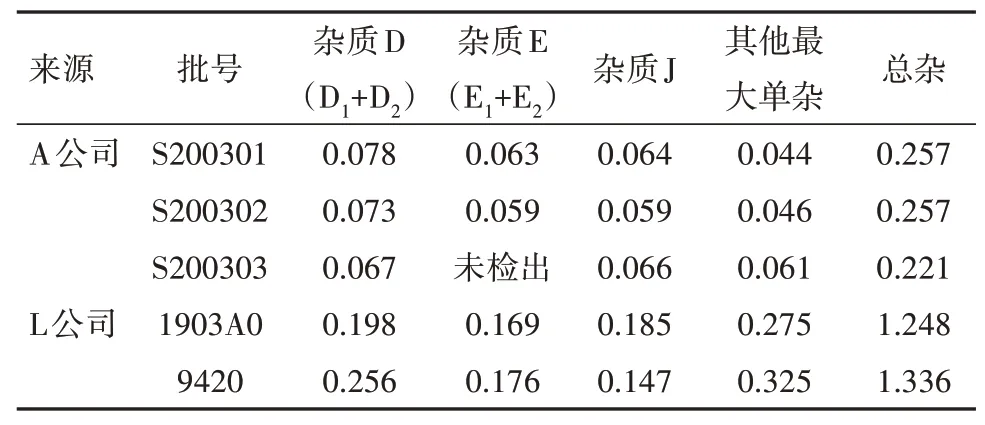
表8 阿莫西林样品测定结果(杂质对照品外标法)/%
由表7,8 的结果可知,分别采用主成分自身对照法加校正因子法和杂质对照品外标法计算,样品中的已知杂质、其他最大单个杂质及总杂质量均差异无统计学意义,校正因子法结果可靠。
2.12 检测波长、流速的选择取同一批阿莫西林颗粒,参照USP41“阿莫西林口服混悬剂”征求意见稿中有关物质检查的色谱条件,分别对210 nm、230 nm、254 nm 三个波长进行了考察,结果见表9。

表9 阿莫西林颗粒不同检测波长试验结果
由试验结果可知,在2.0 mL/min 流速下,230 nm 波长杂质的检出个数及杂质量均比210 nm 和254 nm 波长要多,各杂质间分离度良好,符合要求,耐用性试验中也对流速也进行了考察,故检测波长选择230 nm、流速选择2.0 mL/min 较为合理,且与USP41“阿莫西林口服混悬剂”征求意见稿检测波长一致。
3 讨论
3.1 校正因子的确定由“2.5”项试验结果可知,杂质A、B、D、G、J、K、L、M 的校正因子大于1.1,计算时需加校正因子;杂质F、杂质I、D-对羟基苯甘氨酸甲酯盐酸盐的校正因子小于0.9,计算时采用不加校正因子,相当于严控;其中杂质A、B、G 为原料引入的工艺杂质且样品中未检出,杂质K、L 在强制降解试验中只在液体破坏下产生,未产生杂质M,且本品为固体,故在测定时不再单独控制这6个杂质,也不再加校正因子,按照其他单个杂质控制。
因杂质D、E、J 为本品的主要降解杂质,故在计算杂质含量时,需乘以相应的校正因子。其中,杂质D1校正因子为1.4、杂质D2校正因子为1.4、杂质E1校正因子为1.1、杂质E2校正因子为1.1、杂质J 校正因子为1.2。其他已知杂质均按照其他单个杂质控制,并不再乘以校正因子。
3.2 系统适用性溶液的确定本法中所用阿莫西林系统适用性对照品购自中国食品药品检定研究院,未对具体成分及纯度作出说明,提供的标准色谱图中除标注了阿莫西林色谱峰外,仅标注了阿莫西林杂质C、D、J色谱峰。考虑到杂质D、E、J为本品的主要降解杂质,故在系统适用性试验中,使用阿莫西林系统适用性对照品配制系统适用性溶液1,使用阿莫西林对照品和杂质E对照品配制系统适用性溶液2,分别进行系统适用性试验。
3.3 杂质控制限度的确定现行《中华人民共和国药典》(ChP)2015年版和2020年版中“阿莫西林颗粒”标准中未收录“有关物质”检查项,仅在原料药中对杂质的限度进行了规定。根据现行版《中华人民共和国药典》(ChP)附录[21]中有关的指导原则和国际人用药品注册技术协调会(ICH)Q3B(R2)中对杂质限度的规定,同时结合本品的稳定性试验数据结果,可将本品有关物质限度定为:杂质D(D1+D2)≤2.0 %;杂质E(E1+ E2)≤2.0 %;杂质J≤1.0 %;其他单个杂质≤0.5 %;总杂质≤5.0 %;小于0.01 %的单个杂质忽略不计。
4 结论
阿莫西林颗粒目前仅被中国药典收录,国外药典中的相似剂型为阿莫西林口服混悬剂,被BP、USP 收录;A 公司的阿莫西林颗粒现执行标准为《中华人民共和国药典》(ChP)2020年版二部“阿莫西林颗粒”。本研究在各国药典的基础上,同时按照《化学药物质量控制分析方法验证技术指导原则》《化学药物质量标准建立的规范化过程技术指导原则》《化学药物杂质研究技术指导原则》以及现行版《中华人民共和国药典》(ChP)附录中有关的指导原则,结合本品的制剂特性,对现执行标准进行了优化提高,提出了有关物质的检测方法,为阿莫西林颗粒中杂质谱分析和药品生产过程中的质量控制提供了重要依据。
