基于密度泛函理论的负载型氧化物催化剂CO氧化的研究
2023-06-13陈坤露杜学森刘洋谷郑子文陈艳容
陈坤露,杜学森, *,王 星,刘洋谷,郑子文,陈艳容
(1. 重庆大学 能源与动力工程学院, 重庆 400044; 2. 重庆大学 低品位能源利用技术及系统教育部重点实验室, 重庆 400044)
0 引 言
CO是大气中主要的污染物之一,主要来源于汽车尾气以及燃煤烟气等。CO排放严重影响了人类的健康生活以及社会的可持续发展理念[1-2]。目前,对于CO控制最普遍的方法为催化氧化法[3-5],即在催化剂的作用下,CO和O2发生反应生成CO2。这是非均相催化领域研究最为广泛的反应之一。如何得到高效的催化剂,一直是研究人员们所关注和感兴趣的话题。其中以Au[6-8]为代表的贵金属催化剂由于优异的氧化活性而长期受到人们的青睐,但在当今能源资源越发匮乏的大背景下,贵金属催化剂由于储量有限、价格昂贵等缺点无法广泛应用于工业之中,所以人们逐渐将目光转移到非贵金属氧化物催化剂上。多个研究小组报道的CuO/CeO2[9-11]催化剂,以及其他的Mn[12-13]、Co[14-15]、Ni[16-17]等氧化物,皆对CO具有一定的催化活性。但这些研究仅是对某种催化剂进行研究,缺乏系统性研究。对于结构类似的负载型氧化物催化剂而言,其CO氧化活性是否遵循一定的规律以及我们如何找寻出其中暗含的关系呢?
针对以上问题,我们着眼于当今发展较为成熟、广受关注的计算筛选催化剂的方法[18-19]。此方法是基于密度泛函理论(DFT)对催化反应历程进行模拟计算,从中提炼出能表征催化剂活性的描述符,进而得到活性最佳的催化剂。相较于传统试错形的实验筛选方法而言,这种方法极大程度提高了催化剂筛选的效率并从微观层面上揭示了催化反应的机理,更有助于我们理解和分析催化剂上所发生的反应[20]。目前,已有大量的研究人员使用DFT计算的方法对CO氧化催化剂进行筛选。Han和JW[21]科研团队基于计算对稀土金属掺杂到CeO2催化剂中的CO氧化反应进行了研究,并得出掺杂原子的离子半径可作为CO反应能垒的描述符这一结论。同时通过实验验证了这一结论的合理性、正确性,并预测La掺杂的催化剂将展现出最佳的催化活性。Joseph S. Elias[22]和Bingxian chu[23]等团队也开展了类似的研究,皆表明计算筛选催化剂的高效性及可行性。但对于负载型金属氧化物催化剂上的CO催化剂筛选工作却寥寥无几,特别是以TiO2[4, 24]为载体,过渡金属氧化物为活性组分的催化剂。所以本文将针对这一研究空缺开展相应工作,通过一系列DFT计算得到影响负载型过渡金属氧化物催化剂CO氧化活性的关键。
本文的主要工作是在负载型过渡金属氧化物催化剂表面进行CO氧化反应的分子模拟,详细地分析和讨论各个催化剂表面所发生的反应,得到影响CO氧化活性的关键因素。结果表明,CO在催化剂表面的吸附能(Eads)和活性组分中氧的空位形成能(Eov)大小与催化剂的CO氧化活性密切相关。因此,可以将这两个影响因素作为此类催化剂的描述符,用以评估和预测催化剂的CO氧化活性。为了证明DFT计算的正确性,制备了相应的金属氧化物催化剂,随之开展CO氧化反应性能测试实验,发现实验结果与计算结果相一致。
1 实验方法
1.1 计算细节
本文主要使用Vienna Ab-initio Simulation Package (VASP) 5.4.4和Materials Studio 8.0等工具进行密度泛函理论计算。周期性结构模型采用广义梯度近似(GGA)的PBE泛函来描述电子之间的相关交换能。采用投影缀加平面波(Projector Augmented Wave)赝势来描述原子核与价电子的相互作用,截断能设置为500 eV。对于结构优化,总能量的收敛标准设置为1×10-5eV,每个原子上力的收敛标准设置为0.02 eV/Å;使用了1×1×1 k点的Gamma网格。反应过渡态的计算选择的方法是CI-NEB(Climbing Image Nudged Lastic Band)方法,此时每个原子上力的收敛标准设置为0.05 eV/Å。为了得到更精确的PDOS结果,PDOS计算中K点设置为5×5×3。
我们建立了一个(1×2)、真空层为15 Å的TiO2(101)超晶胞,共48个原子,两层,进行结构优化时固定最下面一层,如图1(a)、(b)所示。本文采用DFT+U的方法来削减含有d轨道原子在计算时由于强库仑力相互作用所造成的误差[25-32]。通过DFT计算,优化得到各个催化剂的稳定构型如图1(c~f)所示,分别代表VO3H、ZrO2、WO3、MnO/TiO2的稳定构型。此外,Cr3OH/TiO2的催化剂理论模型与VO3H/TiO2相同,其余二价负载氧化物催化剂理论模型与MnO/TiO2相同。

注:Ti、O、H、V、Zr、W、Mn分别用浅灰色、红色、白色、深灰色、 青色、蓝色和紫色代表图1 理论计算构型Fig. 1 The theoretical computational configurations
根据以下公式计算催化剂表面的吸附能(Eads)、氧空位形成能(Evf)和反应能垒(Ea)。
Eads=Eslab+adsorbate-Eslab-Eadsorbate
(1)
Evf=Edef-slab+Eo-Eslab
(2)
Ea=ETS-EIS
(3)
式中:Eslab、Eadsorbate、Eslab+adsorbate分别指干净催化剂表面、吸附物和吸附模型优化后的能量;Edef-slab、Eo分别指带有一个氧空位缺陷的催化剂表面能量和一个氧原子的能量(本文通过计算得Eo=-1.89 eV);ETS、EIS分别代表过渡态和初态的能量。
1.2 实验细节
本文所有催化剂均通过传统的等体积浸渍法方法制备[33-34],所有催化剂与质量分数为2%CuO/TiO2保持相同摩尔数。主要制备方法如下:计算并称取不同活性组分所需的量,将活性组分放置于烧杯中并加入一定量的去离子水溶解,随之将烧杯置于磁力搅拌器中搅拌,最后配成一定浓度的前驱液。使用移液枪将配置好的前驱体溶液滴入TiO2载体粉末中,用玻璃棒搅拌直至混合均匀。随后将得到的混合物在室温下放置12 h后放入干燥箱中以110 ℃干燥12 h。接着将干燥后的混合物移入坩埚中,在马弗炉中以500 ℃煅烧5 h。煅烧完毕的混合物经研磨处理后即为实验所需的催化剂。
将制备完成的催化剂研磨并筛选出40~60目大小范围的颗粒进行CO氧化反应性能测试。称取200 mg催化剂置于石英管反应器内,进料气由5 000 ppm CO、10%O2以及平衡气体N2组成,总流速为1 L/min,对应气体空速GHSV为300 000 h-1。温度测试区间为50~400 ℃,在测试时样品至少在测试温度中恒温保持半小时以上以确保反应已经稳定,升温速率为10 ℃/min。出口气体经傅里叶红外烟气分析仪,实时记录反应器出口处CO气体的浓度,其催化剂的CO氧化活性主要由CO转换率(COconversion)来表示,通过公式(4)计算得出:
(4)
式(4)中:COin表示CO在石英管反应器入口浓度,10-6,COout表示CO在石英管反应器出口浓度,10-6。
2 结果和讨论
2.1 CO分子在催化剂表面的吸附
反应物分子在催化剂表面的吸附是反应进行的首要步骤。为了研究CO分子在催化剂表面的吸附对氧化反应的影响。通过DFT计算得到了CO分子吸附的相关信息,包括稳定吸附构型的吸附能,反应物与催化剂表面之间的距离等,具体数值如下表1所示。对于CO而言,其最高占据轨道(HOMO)为5σ,最低占据轨道(LUMO)为2π*。当气相CO在过渡金属氧化物位点进行吸附时,5σ轨道与金属电子未占据态发生杂化,CO分子主要以C端吸附在过渡金属位点。吸附能在-0.12~-0.83 eV之间,其中CO分子在V、Cr和W位点上的吸附能远低于其他过渡金属位点。C端与金属位点之间的键长也存在显著的差异,C端与V、Cr之间的键长长达3 Å,但与其它金属位点之间的键长平均在2 Å左右。

表1 反应物分子CO在MxOyHz/TiO2催化剂表面的 吸附能(Eads),C端与表面之间的距离(dC-M)Table 1 The adsorption energy (Eads) of reactant molecule CO on the surface of MxOyHz/TiO2, and the distance between the C-terminus and the surface (dC-M)
为了深入分析反应物分子CO与催化剂表面的相互作用,我们研究了以上所有稳定吸附构型的态密度(density of states)。以VO3H/TiO2和FeO/TiO2为例,图2是CO分子在催化剂表面吸附前后,其C(up)轨道的态密度图(DOS)和V、Fe的d(up)轨道部分态密度(PDOS)图。由图2可知,CO分子在吸附前,C轨道与V和Fe的d轨道均无重合;吸附后,C轨道与V的d轨道几乎没有变化,但C轨道与Fe的d轨道发生显著变化,且轨道重合。以相同方法分析其余催化剂的态密度图可知,CO分子弱吸附(物理吸附)在V、Cr和W表面,化学吸附在其余过渡金属氧化物表面。
2.2 CO分子在催化剂表面反应的氧化机理
CO分子吸附在催化剂表面后,将与活性组分中的晶格氧进行反应生成CO2,遵循MvK机理[35-36, 1]。随之我们对MxOyHz/TiO2催化剂上的CO催化氧化反应路径进行了分子模拟,其相对反应能量曲线如图3所示。由图3可知,负载型过渡金属氧化物表面上的CO催化氧化过程分为两个反应阶段。第一个阶段为CO与活性组分MxOyHz中的O反应,此阶段由三个基元反应组成。首先,CO分子吸附在催化剂表面的过渡金属位点,作为整个反应的初态(IS1),其稳定吸附构型的吸附能和键长几何参数如表1所示。随后,CO与过渡金属氧化物中的O反应生成CO2(FS1),过渡态记为TS1。最后,反应生成的CO2从催化剂表面脱附,形成一个具有氧空位缺陷的MxOy-1Hz/TiO2催化剂表面E1。第二个阶段包括O2的吸附和CO的氧化。首先,O2在带有氧空位缺陷的催化剂表面吸附形成稳定构型E2,此时还原的MxOy-1Hz/TiO2催化剂被O2再氧化,该构型具有较强的氧化性。随后,CO分子再次被吸附到催化剂表面,与具有较强氧化性的O进行反应生成CO2,此过程同样由三个基元反应组成,与第一个反应阶段相同,即CO的吸附(IS2)、CO2的生成(TS2-FS2)和CO2的脱附(FS2)。最终,催化剂恢复到初始表面,完成催化剂表面的CO氧化循环。
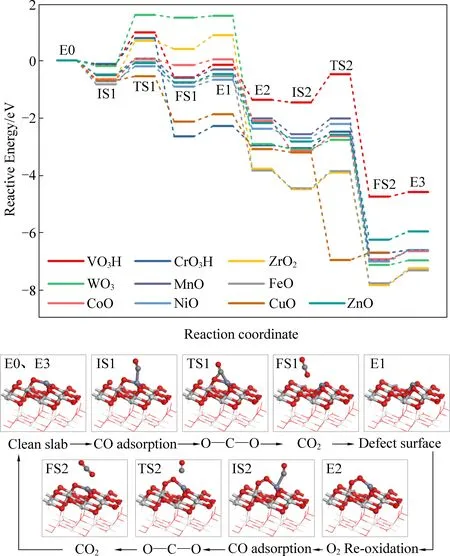
图3 MxOyHz/TiO2催化剂表面进行CO氧化反应的相对能量图Fig. 3 Relative energy diagram of CO oxidation on MxOyHz/TiO2 surface
洞悉所有过负载型渡金属氧化物催化剂表面的CO氧化反应历程,发现CO氧化的关键是从吸附的CO转变为CO2(IS1-TS1,IS2-TS2)的过程,两个反应均为吸热反应,对应的能垒分别为Ea1、Ea2(详细值见表2)。为了比较不同催化剂的CO催化氧化活性,将在这两个反应中选择其中具有最高反应能垒的基本步骤,视为整个反应的速控步。通过比较将基元反应IS1-TS1作为CO催化氧化反应的速控步骤,能垒Ea1则代表催化剂表面发生CO氧化反应的难易程度。同时发现反应速控步的能垒与过渡金属氧化物的氧空位形成能结合以上DFT计算结果,我们将进一步对MxOyHz/TiO2催化剂上的CO氧化反应进行讨论,厘清什么是影响反应能垒Ea1的关键因素。图4(a)为负载型过渡金属氧化物中氧的空位形成能与反应速控步能垒Ea1之间的关系图。由图可知,所有催化剂被划分为两类,其中VO3H、CrO3H、ZrO2、WO3/TiO2催化剂为一类,其余为另一类。在图中两个区域内,氧空位形成能Evf和反应能垒Ea1之间都存在相同的线性关系,即正比关系。由此可得,虽然氧空位形成能可以在一定程度上代表此类催化剂CO氧化能垒的高低,但不是唯一的决定因素。它仅可作为CO氧化活性的描述符之一。结合前文CO分子在催化剂表面的吸附行为,将其作为另一个描述符,两个简单的描述符共同影响MxOyHz/TiO2催化剂的CO氧化活性。图4(b)为速控步能垒Ea1受CO分子吸附能和活性组分氧空位形成能影响的三维云图。由图可知,在这两个因素的制约下,催化剂活性呈现良好的分布规律,颜色越红代表能垒越低,活性越高。由此可得,对于在负载型过渡金属氧化物催化剂上进行CO氧化反应时,只有同时具备较强的CO分子吸附能和较低的氧空位形成能这两个条件时,才能使催化剂具有较高的CO催化氧化活性。在之后的工作中,可以利用这一结论来设计具有高CO氧化活性的催化剂,同时也可以预测其他类似催化剂的CO氧化活性,从而节约大量时间成本。
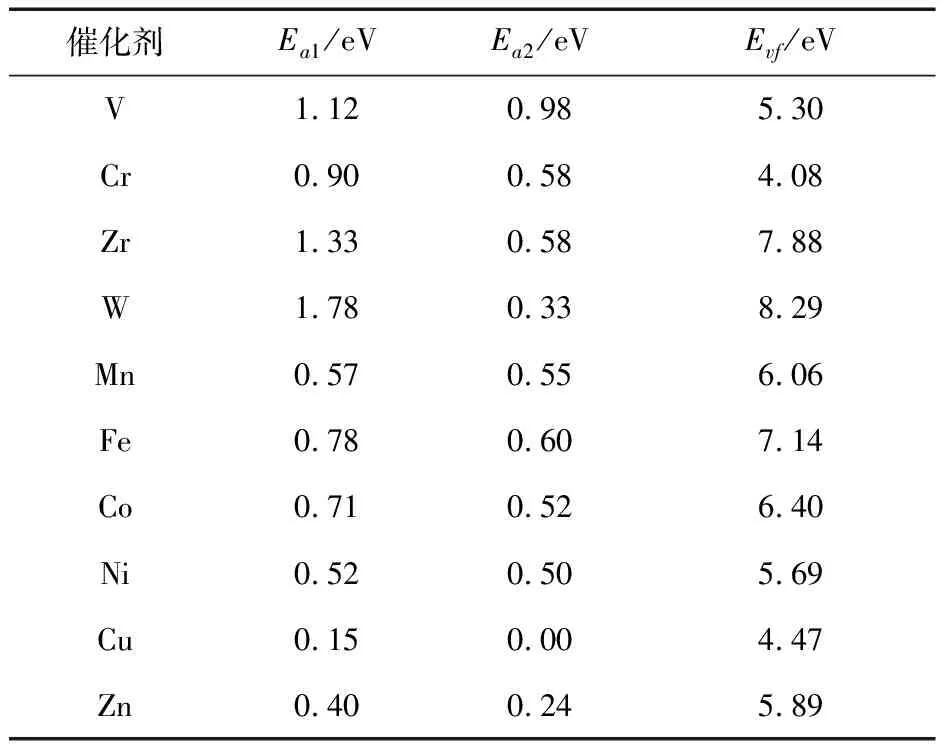
表2 MxOyHz/TiO2催化剂上CO氧化反应能垒 (Ea1,Ea2)和氧空位形成能(Evf)Table 2 The CO reaction energy (Ea1, Ea2) and oxygen vacancy formation energy (Evf) of MxOyHz/TiO2 catalyst

图4 吸附能、氧空位形成能与催化剂CO氧化活性之间的关系Fig. 4 Relationship between adsorption energy, oxygen vacancy formation energy and the CO oxidation activity of catalysts
Evf(详细值见表2)密切相关。
2.3 吸附能(Eads)、氧空位形成能(Evf)与催化剂CO氧化活性的关系
考虑到以上所计算的部分负载型氧化物与实际催化剂的价态不符,主要是Mn、Fe和Co氧化物,随之根据以上得出的结论对这几个负载型过渡金属氧化物真实价态的CO催化活性进行简单计算和预测,以证明该结论的适用性。计算方法同上,通过计算发现MnO2、Fe2O3、Co3O4/TiO2与MnO、FeO、CoO/TiO2相比,其各自的活性都高于之前二价状态的氧化物,但都比CuO/TiO2的活性低。基于此,通过所有的计算得到了以上负载型过渡金属氧化物催化剂的CO催化活性,并将在下一节对计算结果进行验证。
3 活性验证
通过在负载型过渡金属氧化物催化剂表面进行CO氧化分子模拟,我们得到CuO/TiO2催化剂的活化能垒最低,CO催化氧化的活性最高;相反VO3H、CrOH3、ZrO2、WO3/TiO2的活化能垒最高,活性最低;其余催化剂的活性介于两者之间。随后我们制备了相应的金属氧化物型催化剂,并开展了CO氧化反应性能测试实验,发现实验结果与DFT计算结果一致,这为DFT分子模拟的正确性提供了有力的证据。
图5展示了不同活性组分的负载型氧化物催化剂的CO转化率随时间的变化曲线。所有催化剂中,CuO/TiO2催化剂展现出最佳的CO催化氧化性能,在280 ℃时,CO转化率达到100%并保持不变。V2O5、ZrO2/TiO2两者的催化活性最差,在反应温度区间内基本没有CO转化,当反应温度达到400 ℃时,转化率仅为5%。其余催化剂的CO催化氧化活性相当,但在反应结束时,最高的CO转化率都低于50%。以上实验结果与DFT计算结果相吻合,说明了DFT计算的正确性。
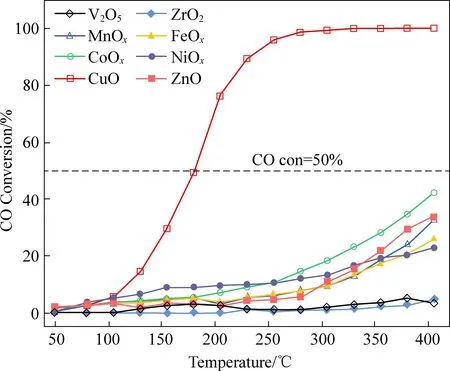
图5 不同的负载型氧化物(MxOy/TiO2)催化剂 CO转换率随温度的变化曲线Fig. 5 Dependence of CO conversion on reaction temperature with different supported oxide catalysts (MxOy/TiO2)
4 结 论
本文在一系列负载型过渡金属氧化物催化剂表面进行了CO氧化分子模拟。随后我们通过分析得到影响催化剂表面进行CO催化氧化反应的关键因素,即CO在催化剂表面的吸附能(Eads)和活性组分中氧的空位形成能(Eov)。通过讨论发现这两个简单的因素可以作为此类催化剂CO催化氧化活性的描述符。随后我们便利用以上结论对MnO2、Fe2O3、Co3O4/TiO2的CO活性进行简单计算和预测。结果得到在所有催化剂中,CuO/TiO2催化剂CO氧化的活性最高;相反VO3H、CrOH3、ZrO2、WO3/TiO2的最低;其余催化剂的活性介于两者之间。最后我们制备了相应的金属氧化物型催化剂,并开展了CO氧化反应性能测试实验,发现实验结果与DFT计算结果一致,这为DFT分子模拟的正确性提供了有力的证据。同时也证明CO分子的吸附能和活性组分中的氧空位形成能可以作为CO氧化活性的描述符,这为我们之后进一步的催化剂筛选以及设计工作提供明确方法。
