机器学习加速能源环境催化材料的创新研究
2023-06-13林赛赛傅雨杰赵海涛刘少俊张涌新郑成航
张 霄, 董 毅, 林赛赛, 傅雨杰, 徐 丽, 赵海涛, 杨 洋,刘 鹏,刘少俊, 张涌新,郑成航, 3,高 翔, 3, *
(1. 浙江大学 能源高效清洁利用全国重点实验室, 浙江 杭州 310027;2. 浙江大学 碳中和研究院, 浙江 杭州 310027;3. 白马湖实验室, 浙江 杭州 310051)
0 引 言
“十四五”时期我国进入新的发展阶段,加快推动能源的清洁高效利用与生态环境的改善治理已成为破解资源环境约束、实现可持续发展的迫切需求[1-2]。催化剂作为能源化学与环境化学的重要媒介,在推动化石能源清洁高效利用[3]、可再生能源规模化应用[4]以及二氧化碳捕集与利用[5]等方向的技术发展起到了关键作用。近年来,电催化、热催化与光催化技术在制取绿氢、合成绿色燃料与提升燃料电池效率等能源领域迎来了重大发展机遇[6-8],也为温室效应以及大气污染等环境问题提供了有效解决途径。因此,催化材料在能源与环境领域中显现出了广阔的应用前景。
开发有助于清洁能源高效生产及污染物脱除或循环利用的新型催化材料是解决现有能源与环境问题的关键。目前,催化材料的设计已经从组分调节、尺寸控制等宏观调控延伸到了基于原子/分子水平的精准调控[9],如催化中心配位结构和电子结构调控。以经验指导为主的传统催化剂研发模式由于筛选效率低、开发周期长和易陷入局部最优解等缺点,严重制约了新型催化材料的研发[10]。数据科学的快速发展为先进催化材料的研发带来了新的思维模式和技术路径[11]。通过有机结合催化实验、基础物理/化学理论、计算模拟和数据技术,可加速先进催化材料研发、催化反应机理揭示以及催化反应性能识别与催化活性位点预测。这种数据科学结合先验知识的机器学习研究方法,旨在从数据中提取尚未掌握的知识进而指导开展催化剂研究[12],加快催化剂构效关系的建立、实现催化剂的按需设计以及催化活性位点的定向构建,降低研究难度和开发成本,推动催化材料研发由“经验+试错”模式向“知识+数据”融合模式转变。
通过对机器学习方法在催化材料中的研究热点进行聚类分析(图1),可以看到机器学习可对催化材料研发的各个方向进行加速和优化,逐步从催化剂配方的快速筛选、催化材料形貌结构的理性设计深入到对微观催化活性位点的准确预测以及催化反应机理的清晰揭示。因此,本文将从催化材料的位点预测、配方筛选、构型设计以及路径优化等方面,综述机器学习方法在能源与环境催化材料研发中的相关研究进展,重点分析基于不同训练数据来源所对应的机器学习方法构建以及应用方向,从而为机器学习方法在能源与环境领域的进一步应用提供理论和技术启发。

图1 机器学习辅助催化材料研究的关键词共现聚类分析可视化知识图谱 (数据来源与处理:以“machine learning” and “catalysis”为关键词在Web of Science核心合集中检索2012-2023年间 的所有文献;采用VOSviewer软件进行可视化聚类分析)Fig. 1 The results of cluster analysis coupled with word frequency statistics of keywords in all the publications with “machine learning” and “catalysis” as topics obtained from Web of Science over the past 10 years (conducted by VOSviewer)
1 位点预测
精准识别催化活性位点是理解催化机制和合理设计催化剂的基础。通过计算模拟可以得到催化活性位点原子的配位环境、电子结构、催化反应路径、过渡态、中间体以及吸附能等信息,利用这些数据可以分析影响催化性能的关键反应中间体和速控反应步骤等[13-14]。将机器学习与计算模拟相结合,可在只计算部分催化活性位点样本相关信息的基础上预测大量的催化活性位点性能,极大地降低了计算资源需求,加速催化材料的预测和设计,缩短研发周期,降低研发成本。
密度泛函理论(Density Functional Theory,DFT)是研究催化剂表面催化活性的重要理论基础[13]。为了解决密度泛函理论方法在计算大量多原子体系时计算成本高等难题,Wan等人[15]开发了一种机器学习加速的密度泛函理论计算(DFT-ML)方法,用于预测17种过渡金属元素的单金属及双金属中心酞菁催化剂的电催化CO2还原性能(图2)。作者首先使用DFT方法计算了17种单金属中心以及40种随机组合双金属中心催化剂的CO2还原各基元反应自由能变,以此获得催化剂的极限电位来表征其催化性能。在此基础上,建立包含催化剂中心金属原子半径、d轨道电子数、电负性等特征值及催化性能的数据库用于机器学习模型训练,并使用训练后的机器学习模型预测了另外249种双金属中心酞菁催化剂的催化性能,从而对研究中所包含的单金属及双金属中心酞菁催化剂性能的预测效率提高了约7倍。
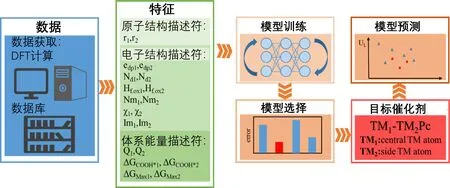
图2 机器学习结合密度泛函理论预测催化活性位点工作流程图[15]Fig. 2 Flow diagram of accelerated catalytic activity prediction by combining machine learning with density functional theory[15]
基于对反应机理的深入认识,通过计算少量关键中间体吸附能等信息获得催化剂的性能,从而避免计算完整反应路径中各基元反应的能量变化,可进一步提高机器学习与计算模拟结合方法预测催化活性位点性能的效率[16]。例如,CO是CO2还原反应的关键中间体[17],其吸附能常被用于预测催化剂的CO2还原反应性能[18]。Zhong等人[5]从Materials Project数据库中获取了244种含Cu金属间化合物晶体结构,据此列举了12 229个晶面和228 969个活性位点,然后通过DFT计算方法获取了其中部分活性位点样本的CO吸附能作为机器学习预测目标值,并以活性位点元素的原子序数、电负性、单金属晶面CO吸附能中值等作为特征值进行机器学习模型训练,再由此获得所列举的其他活性位点的CO吸附能,以此预测具有高CO2还原反应活性和多碳产物选择性的Cu基催化剂(图3(a))。Xing等人[19]同样从Materials Project数据库中获得了铜基合金的结构,并以CO、COOH和HCOO等不同中间体吸附能作为机器学习目标值预测了生成不同CO2还原产物的铜基合金催化剂。

图3 机器学习与计算模拟结合预测催化活性位点Fig. 3 Prediction of catalytic active sites by combining ML and computational simulation
机器学习与计算模拟结合方法也被用于高熵合金(High Entropy Alloys, HEAs)等其他体系催化剂的高效预测[21-23]。Pedersen等人[20]使用CoCuGaNiZn和AgAuCuPdPt高熵合金体系中活性位点中心原子的配位环境等特征以及DFT计算所得的反应中间体吸附能等数据训练机器学习模型,以此预测了所有可能活性位点的CO和H吸附能,最终获得了不同元素比例HEAs催化剂的CO2/CO还原反应活性及选择性(图3(b))。Roy等人[24]也通过机器学习与计算模拟结合方法筛选了用于CO2加氢制甲醇的HEA催化剂体系,并获得了35种高活性、高选择性的CO2加氢制甲醇催化剂(图3(c))。
由于实际催化剂颗粒中存在位于不同晶面、边缘、缺陷等多种活性位点,机器学习与计算模拟结合方法还可以用于识别单个催化剂纳米颗粒中的高性能催化活性位点,以此指导催化剂的可控构筑。Chen等人[25]首先采用分子动力学模拟构建了一个置于碳纳米管之上的粒径为10 nm的金催化剂颗粒,其中包含了11 537个表面金位点,然后依据表面原子结构提取结构描述符,与量子化学计算所获得的部分表面金位点CO吸附能及HOCO中间体生成能相结合构建机器学习模型训练数据库,以此预测所有表面金原子的催化性能,实现了整个金催化剂纳米颗粒表面原子催化活性位点的可视化(图4)。
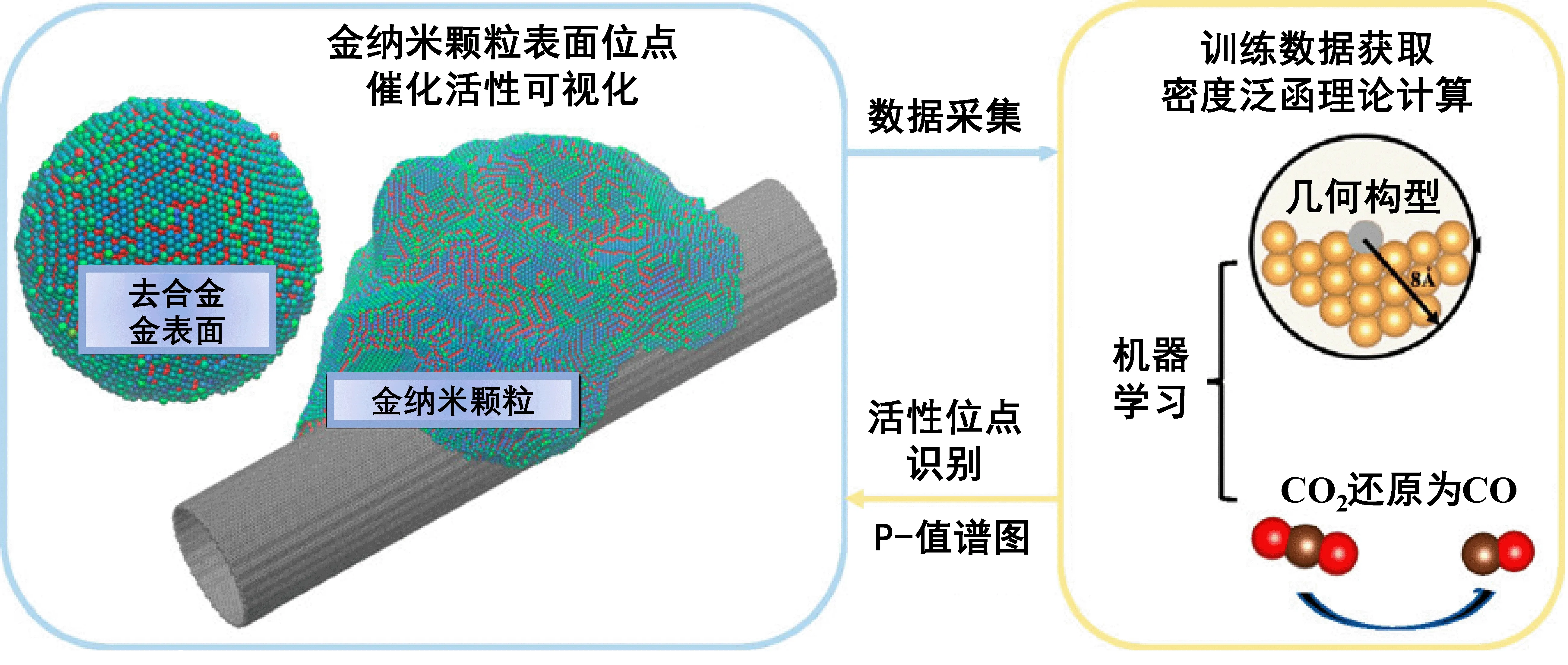
图4 机器学习结合多尺度模拟可视化去合金表面二氧化碳还原活性位点[25]Fig. 4 Visually identifying active sites for CO2 reduction on dealloyed gold surfaces by combining machine learning with multiscale simulations[25]
2 配方筛选
催化剂组成元素及含量、制备方法及条件等多种配方参数与催化剂活性、选择性、稳定性、抗中毒能力等多种性能指标之间存在强相关性[3]。文献调查[26]、理论计算[27]和高通量实验[28]等方法是获取催化剂相关数据的重要途径,机器学习可结合不同途径所得数据指导相关能源与环境应用场景需求下的新型催化剂筛选、制备及反应条件优化。
选择性催化还原(Selective Catalytic Reduction,SCR)是减少氮氧化物(NOx)排放的有效方法,在催化剂的作用下NOx和还原剂NH3反应生成无害的N2和H2O。高翔等人[3]构建了一种典型的基于文献调查数据的机器学习预测方法,用于快速筛选低温SCR催化剂配方以及优化反应条件(图5)。该方法主要分为四个步骤:(1)基于2 000余篇已发表的相关文献构建了低温SCR催化剂组成-性能信息数据库,并对数据进行预处理;(2)选择金属氧化物作为主要研究对象,构建了由催化剂组成、结构等20余个特征变量信息组成的催化剂特征向量;(3)通过对比不同机器学习回归预测模型的精度,筛选出具有最优预测精度的极端随机树回归模型;(4)利用训练好的机器学习模型筛选获得了具有良好低温脱硝性能的Mn-Ce-M(M=Co、Cu、Fe)催化剂体系。Chen等人[29]采用相似的机器学习与文献调查相结合的方法,利用遗传算法实现了MnTi催化剂元素配比及制备条件的优化。相较于上述的金属氧化物SCR催化剂,沸石类SCR催化剂多用于柴油车后处理等移动源催化脱硝场景中[30]。Bae等人[31]以沸石基底的硅铝比、金属组分及含量、制备方法及反应条件等多种特征参数构建特征向量,并以NOx转化率作为催化剂的性能指标,将数据库按照沸石类型划分为β型和ZSM-5型催化剂两个独立数据子集进行训练,使用递归式特征消除(Recursive Feature Elimination,RFE)算法从原始特征逐步迭代从而生成机器学习预测模型[32]。上述方法也被应用于CO氧化[33]、CO2加氢制甲醇[34]、甲烷干重整[35]等反应中,并通过模型筛选实现了催化剂配方及反应条件的优化。
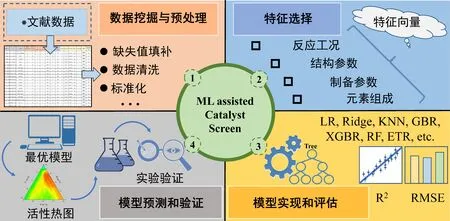
图5 文献研究数据驱动的机器学习开发催化材料工作流程示意图[3]Fig. 5 Schematic diagram of machine learning aided catalyst optimization based on literature research data[3]
贵金属催化剂(如铂、钯等)因其高活性和选择性常被用于燃料电池、电解水产氢系统和空气电池等能源领域[36]。受限于地球储量稀少(如铂地壳含量仅为亿分之五)、成本高昂(如铂200-250元/g),如何提高贵金属原子的利用效率成为贵金属催化剂开发的核心问题之一[37-38]。通过机器学习与理论计算相结合,可以低成本、高精度地实现单原子或复杂原子催化剂体系的快速筛选与合成调控。黄勃龙等人采用机器学习与理论计算相结合的途径构建了一种氧化还原势垒模型,通过量化描述d/f轨道电荷转移,实现了对零价原子催化剂锚定的“石墨炔-原子催化剂”组合体系的快速筛选[39],并借助机器学习与理论计算相结合的途径,对石墨炔基HER原子催化剂进行了全过渡、稀土金属的有效筛选[40],进一步佐证了理论计算结合机器学习定向筛选原子催化剂体系的可行性。
相较于文献报道数据存在碎片化和标准格式不统一、理论计算数据容易引入难以消除的系统误差等限制,自动化高通量实验技术能够高效、连续地产生可重复性数据,更好地满足了机器学习对可用数据的数量、质量和多样性的高要求,受到越来越多研究者的关注。高通量技术用于催化研究过程如图6(a)所示[41],主要包括(1)实验设计;(2)手动或自动制备反应物;(3)构建能够良好控制制备条件(例如温度和反应时间等)的反应器;(4)催化剂的平行表征。获取的高通量实验数据进一步结合机器学习算法自动反馈指导新实验条件参数的设计,从而加速催化剂组成、制备方法以及反应条件等关键参数的优化[42]。自动化高通量实验技术的核心是高通量制备与高通量表征。基于高通量制备实验平台可实现不同种类催化剂的制备策略,如溶液喷涂结合水热反应方法常被用于BaxSryCaZTiO3基钙钛矿纳米粒子等高效氧还原(ORR)催化剂的快速筛选[43];超声喷雾热解技术被用于在导电衬底大批量沉积三元Fe-Co-Mn氧化物催化剂[44];高通量成分梯度溅射法被用于制备析氧/析氢反应的Ni-Mo双金属合金薄膜催化剂[45];热冲击法被用于高熵合金纳米颗粒的快速合成[46]。在高通量表征方面常用单通道自动按序表征和多通道同步表征两种模式。单通道表征一般采用可移动的扫描探头对催化剂阵列的各项参数进行表征记录[47],多通道表征一般采用多通道信号获取设备进行多通道同步表征。两者均可快速表征不同化学成分催化剂的性能[48]。总体而言,针对具体的催化剂反应体系,在综合考量实验效率和实验成本的前提下,对高通量制备方法和高通量表征手段实现系统性地匹配优化设计是开展高通量实验研究的前提。

图6 高通量技术与“机器人化学家”Fig. 6 High-throughput technique and robotic chemist
自动化高通量实验技术和人工智能的融合也催生了以机器化学家为代表的机器科学家的诞生。例如,Cooper等人[42]通过将高通量实验与机器学习算法耦合建立了“机器人化学家”平台(图6(b)),将其应用于光催化水分解制氢催化剂及反应条件的筛选。该方法使用高精度自动机器人在多种反应条件下对系列催化剂进行批量化测试,基于前一轮实验的结果,利用贝叶斯优化算法自主选择实验参数的优化方向。在筛选过程中,算法对已有数据分析后自主制定下一步筛选策略,在短时间内将光催化水分解制氢性能提高了5倍。赵海涛等人[49]研制了由机器学习、机器人自动化和大数据共同组成的“机器人平台”(图6(c)),验证了从化学原料取样、机器人辅助合成、机器人原位表征到机器人逆向设计材料的全过程,构建了纳米金晶、双钙钛矿纳米晶形貌与配方间的关联机制。这种将高通量实验与机器学习结合的方法还被应用于甲烷氧化偶联反应[50-51]及光催化析氧[52]等多种能源与环境催化材料的研发中。
3 构型设计
准确的催化剂构效关系是实现催化材料形貌结构理性设计的基础[53]。原位反应下催化材料的动态结构形成和演化复杂,厘清催化剂的构效关系在实践中具有挑战性。在传统的催化剂研发范式中,由于理论模型的构建往往存在很多近似性假设,所以很难在实验制备过程中完美地复现所设计的结构。此外,受限于现有表征手段的精确性和灵敏性不足,对催化剂表面结构进行全面地、高精准度地表征也存在一定的困难。如图7所示,数据机器学习技术的发展使得研究人员可以基于先验知识和积累的有效数据,开展催化材料动态形貌结构的预测以及反应条件和微环境下的原位模拟[54],从而指导研发具有最佳形貌结构的催化材料。
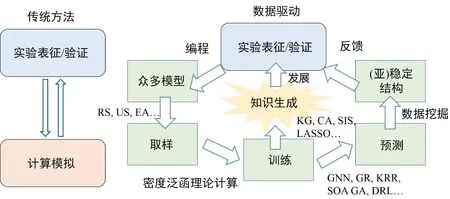
图7 基于传统方法和数据驱动方法的催化剂形貌结构设计工作流程[54]Fig. 7 Workflows for conventional and data-driven methods to design morphology and structure of catalysts[54]
机器学习方法结合人工智能,可以减少人工工作成分,提高实验设计效率,从而改变研究工作流程,允许理论模型直接来自实验发现。这将弥合实验和理论之间的“材料差距”,加深对催化剂形貌结构的理解。研究人员已经开发了自动识别图像的机器学习应用程序,通过与生成图像的实验表征相结合,给出自动化的结构识别,从而节省人力和减少人为错误。Krull等人[55]研发了基于机器学习的人工智能框架(DeepSPM),能够自主连续采集原子精密扫描探针显微镜(SPM)图像(图8(a))。DeepSPM能够在多天的扫描隧道显微镜实验中连续采集和分类数据,选择高质量成像样本区域并执行测量的算法命令,并根据不同实验条件管理探针质量。Girod等人[56]使用冷冻透射电子断层成像方法研究了质子交换膜燃料电池中催化剂层的形貌,并利用深度学习方法辅助完成了图像复原与组分鉴别。作者以此计算了离聚物的形貌、覆盖率和均匀性以及铂催化剂在碳载体上的分布等指标,进而建立了催化剂形貌及传输特性与燃料电池性能之间的构效关系。
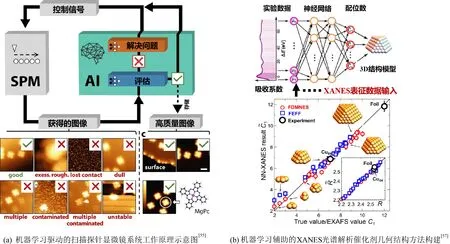
图8 机器学习驱动的催化剂形貌结构信息提取Fig. 8 ML-driven information extraction from morphology and structure of catalysts
机器学习方法还可以高效地从催化剂形貌结构表征所获得的波谱数据中提取信息。例如,X射线吸收谱(XAS)能够原位检测和分析金属纳米粒子(NP)结构与其特性(如催化活性)之间的相关性,这对实现高性能催化剂的合理设计具有重要价值。XAS谱的X射线吸收近边结构(XANES)部分主要反映原子内层电子向未占据空轨道的跃迁,因此对原子的3D排列细节敏感,具有对结构和电子特征的敏感性。阻碍XANES用于纳米催化剂定量分析的主要挑战是缺乏能够从光谱中提取结构特征的方法。Timoshenko等人[57]利用人工神经网络揭示了XANES特征和催化剂几何结构之间的隐藏关系,并使用XANES光谱和监督机器学习完善了金属催化剂的三维几何结构。该方法使得从其实验XANES中求解金属催化剂结构成为可能,对于催化反应过程中催化剂结构的原位变化追踪具有重要指导意义(图8(b))。
4 路径优化
深入理解催化反应机制与路径是设计高效催化剂的关键。传统上,研究人员常基于实验数据构建微观动力学模型来阐述反应机理,然而,速率定律的推导及其解释需要大量的数学近似,容易出现人为错误,并且仅限于稳态近似下由几个基元反应步骤构成的反应网络。基于机器学习的新理论方法为揭示并优化复杂催化体系的反应机理提供了新的途径。
乙烯在银基催化剂上的环氧化反应是一种重要的多相催化反应,围绕环氧化反应机理与路径的揭示一直存在争议。刘智攀等人借助机器学习方法针对此类多相催化反应机理探索开展了大量工作[58-59]。该团队首先采用随机表面行走反应抽样(SSW-RS)方法寻找Ag(100)上的金属氧杂环(OMC)中间体,该中间体是乙烯环氧化的关键中间体。研究发现银金属表面上的乙烯氧化有三条低能垒反应途径,其中最重要的一个是以前完全忽略的羰基金属环中间体(OMC-DH)的脱氢,如图9(a)所示。通过计算自由能曲线和微观动力学模拟,表明无论反应条件如何,脱氢途径对于Ag(100)和Ag(111)金属表面上的乙烯氧化总是占主导地位(> 90%)。该团队还开发了一个用于多相催化体系活性预测的端到端人工智能框架(AI-Cat方法)[59]。如图9(a)所示,简单输入分子和金属催化剂名称,可以推算出从输入分子到低能垒途径产物的反应能量分布。AI-Cat方法结合了两个神经网络模型,一个用于预测反应模式(R-Pat unit),另一个用于提供反应势垒和能量(K-Info unit),利用蒙特卡罗树搜索反应网络中的低能垒路径。该方法被成功应用于铜表面甘油氢解反应网络的解析。
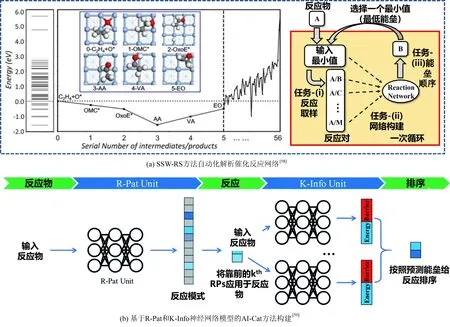
图9 机器学习辅助的银基催化剂上乙烯环氧化反应机理探究Fig. 9 Machine learning assisted reaction mechanism exploration of ethylene epoxidation on silver-based catalyst
涉及到碳氢化合物的反应网络往往由于繁多的物种变化而显得十分复杂。Zachary等人[60]提出了一个使用高斯过程回归(GPR)的反应网络优化框架,以研究在实验相关的操作条件(573 K和1大气压的气相反应物)下,合成气(CO + H2)在Rh(111)催化剂上的反应。通过量子化学计算的部分中间物质的吸附能训练GPR模型,从而预测反应网络中所有中间产物的自由能,结合过渡态比例关系和一个简单的分类器来确定反应的速控步骤。通过将该方法应用到合成气在铑(111)上的反应中确定了其最可能的反应机制。Jordi等人[61]证明了一个基于模拟动力学数据训练的深度学习模型能够正确地阐明来自时间浓度分布的各种机理。训练后的模型可以用来分析普通的动力学数据,并依照训练内容将其按照机理类型分类,在这样一个强大工具的辅助下,动力学分析将由以前冗长繁杂的动力学模型分析和推导简化为自动化的准确预测(图10),增强了合成化学家研究反应机理的能力。
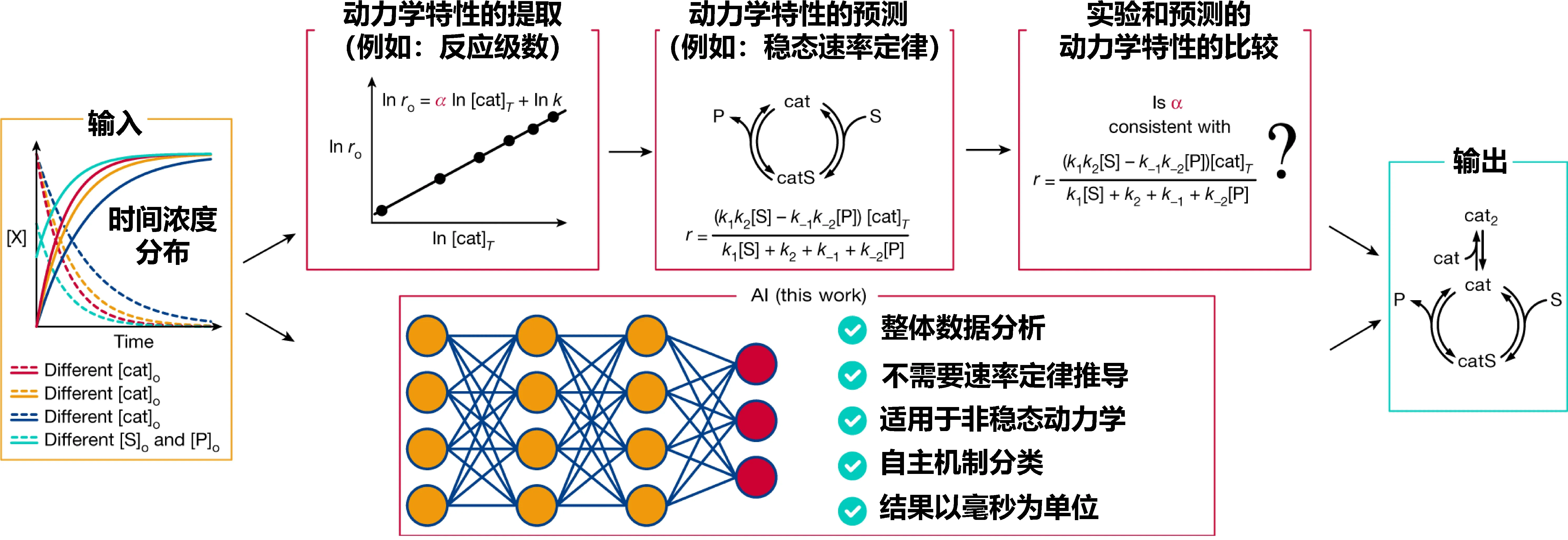
图10 基于传统动力学模型和机器学习解析机制的碳氢催化反应网络研究路径比较[60]Fig. 10 Comparison of current pipeline for mechanistic elucidation through kinetic analysis versus the use of AI-based mechanistic elucidation[60]
5 结论与展望
数据驱动的催化材料研究方法可以将实验、物理/化学理论、计算模拟和数据技术有机结合以指导催化材料的快速设计与优化研究。本文从位点预测、配方筛选、构型设计以及路径优化等角度讨论了机器学习方法在能源与环境催化材料领域的相关研究进展,分析了文献调查、计算模拟、高通量实验等不同训练数据获取途径对应的机器学习方法及其适用的催化材料研究应用方向。使用机器学习加速催化材料创新的研究范式已在寻找潜在高活性催化剂组分、分析影响催化性能的关键因素等方面展现出独特价值,有望成为高性能催化材料开发的重要途径。
在能源与环境催化研究领域,机器学习辅助催化材料开发方法正向着更高效率、更高精度发展,主要方向包括:(1)提高数据收集的效率:开发面向催化研究的ChatGPT类自然语言处理算法,从文字和图表中快速读取数据以建立数据库,并且在催化相关研究工作发表时采用统一的、标准的数据呈现方法,以便快速完成文献数据收集;(2)深入理解催化反应机理:在能源与环境催化反应体系中建立计算模拟结果和催化活性位点性能之间更准确的关联,并且开发结合机器学习的高通量智能计算方法,进一步提高机器学习与计算模拟结合方法在新型能源与环境催化材料研究方面的适用性;(3)开发面向多种反应体系的自动化高通量催化剂制备与测试表征平台:面向包括气-固、液-固等非均相能源与环境催化反应体系,建立自动化、流程化的催化剂制备与评价解决方案,并且进一步挖掘可方便测量的光学、电学等性质以代表催化剂性能,逐步实现基于“机器科学家”的能源与环境催化材料的高效筛选。
