Zn(H2PO4)2/AC在气固相乙炔水合反应中的应用
2023-06-12陈真周龙王庆乐王琴琴代斌
陈真,周龙,王庆乐,王琴琴,代斌
(石河子大学化学化工学院,新疆 石河子 832003)
乙醛是一种重要的化工原料,可作为多种化工产品的中间体,随着工业的不断发展,未来对乙醛的需求量将会不断增加[1-3]。近年来,由于石油资源的日益消耗,原材料丰富的乙炔水合制乙醛工艺逐渐受到国内外学者的广泛关注。对于煤炭资源丰富的国家而言,乙炔法生产乙醛的工艺路线仍具有很高的研究价值[4]。乙炔法又可分为液相和气固相两种,液相法最初采用的是HgCl2的盐溶液作为催化剂,但存在活性低,催化剂难回收等问题,因此气固相反应体系更适合乙炔水合反应。
乙炔水合反应多采用如汞、镉、金、银、铜、锌等过渡金属作为活性组分。汞和镉都属于剧毒金属,会严重危害人体健康和生态环境。金和银都属于贵金属,并且容易被还原为金属单质而失去活性,而铜离子也容易被还原成铜单质,因此,来源丰富、价格低廉的锌金属更适合于气固相乙炔水合反应。Yang等人[5]在Cu+中加入不同的含S配体作为催化体系催化液相乙炔水合反应,实验结果表明,硫代苹果酸的加入显著的提高了乙炔的转化率和乙醛的选择性。Zhang等人[6]通过浸渍法引入HEDP配体,成功地抑制了乙炔水合过程中Cu离子的还原。此外,Li等人[7]结合密度泛函理论,报道了一种S、N共掺杂石墨烯的单原子催化剂ZnN4S2-SAC,该催化剂在吸附反应物后改变了乙炔的电子结构,并促进了水合反应。Li等人[8]合成了N和P共掺杂的Cu基催化剂,大大增加了活性成分Cu的分散性,同时抑制了高价铜的还原。当N和P的掺杂浓度分别为0.4 wt%和2.61 wt%时,乙炔转化率达到90%。Li等人[9]通过密度泛函理论(DFT)方法系统地研究了ZnCl2、Zn(OH)Cl和Zn(OH)2催化剂上此类反应的机制。他们发现,打破水的O—H键所需的能量决定了催化过程的活化能。Wang等人[10-11]发现,使用氨基(APTES、PEI)修饰的介孔MCM-41载体可以有效改善催化剂的分散性,增强金属-载体的作用力,提高活性Zn成分周围的电子云密度,从而改善Zn基催化剂的催化性能。随后,对Zr改性的ZSM-5沸石的研究发现,增加弱酸位点和活性位点的浓度可以提升水合催化活性,乙炔转化率达到96%以上[12]。Wang等人[13]研究了双金属催化剂,发现铜和锌的协同作用能够抑制Zn物种的损失,从而使乙炔转化率高达98%。Wang等人[14]尝试将锌浸渍到Ti掺杂的MCM-41载体上,有效地提升了粒度控制,阻碍了碳沉积。
本文分别以ZnCl2,ZnO作为锌源,活性碳为载体,采用过体积浸渍法制备了Zn/AC、Zn(H2PO4)2/AC催化剂,并在一定反应条件下用于气固相乙炔水合制乙醛反应。对反应前后的催化剂进行了BET,XRD,Py-FTIR,XPS,TG等表征,以分析催化剂的物理化学性质,进而得出催化性能提升的原因。
1 材料与方法
1.1 实验材料
AC(40-60目)为椰壳活性炭,由唐山联合炭业科技有限公司提供;氧化锌(ZnO,分析纯)、磷酸(H3PO4,98 wt%)、氯化锌(ZnCl2,98%),均购买自上海麦克林生化有限公司;高纯氮气(99.99%),乙炔气(98%)均由上海伟创气体有限公司提供。
1.2 催化剂合成
所有催化剂均采用过体积浸渍法制备,其中Zn2+的负载量均为7.2 wt%,具体制备方法如下:
Zn/AC催化剂:用烧杯取20 mL去离子水,加入0.529 4 g商购的ZnCl2,搅拌0.5 h,待固体充分溶解后,加入3 g活性炭载体,继续搅拌12 h后,置于80 ℃的鼓风干燥箱中干燥12 h。
Zn(H2PO4)2/AC催化剂:取30 mL去离子水于烧杯中,加入3 g磷酸(85 wt%),不断加热搅拌,待上述溶液达到120 ℃左右,边搅拌边少量分批次加入0.627 2 g的ZnO,整个过程需要确保溶液始终保持澄清。0.5 h后加入AC,停止加热,继续搅拌12 h,置于80 ℃的鼓风干燥箱中干燥12 h。
1.3 反应催化剂性能评价
乙炔水合反应在自制的固定床反应器(Ø10×500 mm)中进行,将2 mL催化剂样品放置反应管中部。先用N2吹扫30 min,将反应管内的空气除去。然后开始加热,同时设置冷凝循环泵温度约-2 ℃。待反应管升温至240 ℃、反应水蒸汽温度达到160 ℃,通过蠕动泵装置对催化剂进行水汽活化(约0.06 g·min-1,30 min)。然后关闭N2阀门,通入C2H2气体,根据GHSV(C2H2)=90 h-1和n(H2O)/n(C2H2)=4的反应条件,将乙炔和水蒸气注入反应器。最后每隔1 h,通过对气相色谱(GC-2014C)手动进样来检测产物含量。评价指标如下:
(1)
(2)
其中XC2H2为乙炔的转化率,SCH3CHO为乙醛的选择性;ØA0表示初始反应物中乙炔的体积分数,ØA表示产物中残留的乙炔的体积分数,ØAA表示产物中乙醛的体积分数。
1.4 表征测试仪器
比表面积及孔容孔径分析(BET)采用的是麦克公司生产的ASAP-2020C仪器;X射线粉末衍射(XRD)采用德国布鲁克D8 ADVANCE型X射线衍射仪;热重分析(TGA)是德国NETZSCH公司的STA449F3型号的同步热分析仪;X射线光电子能谱(XPS)采用由日本岛津公司生产的Axis Ultra光谱仪;Py-FTIR表征分析采用的是德国TENSOR 27 Bruker红外光谱仪。
2 结果与讨论
2.1 催化性能测试
首先,在240 ℃,GHSV(C2H2)=90 h-1,n(H2O)/n(C2H2)=4的反应条件下,探究了不同Zn基催化剂对乙炔水合反应性能的影响,实验结果如图1所示。从图1a可以看出,Zn/AC催化剂在乙炔水合反应中乙炔的转化率是逐渐升高的,然而反应4 h后,乙炔的转化率迅速降低,这表明活性位点在减少,催化剂可能逐渐发生了失活。Zn(H2PO4)2/AC催化剂中乙炔的转化率是逐渐上升的,10 h后仍保持在90%以上。此外,从图1b可以看出乙醛的初始选择性为53%左右,随着反应的进行选择性逐渐降低,6 h后基本稳定在27%左右,在对反应产物检测分析后,发现丙酮和丁烯醛是该反应的主要副产物。Zn(H2PO4)2/AC催化剂性能最好,更稳定,在反应10 h后,乙醛选择性仍有70%左右。因此,猜测磷酸在乙炔水合反应起到了促进作用。
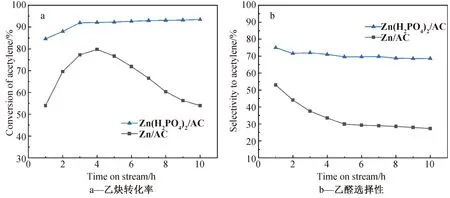
图1 不同Zn基催化剂的活性测试
2.2 BET表征分析
为了探究催化剂的物理结构,对催化剂进行了BET表征分析。从图2a和图2b显示的所有催化剂的氮气吸脱附等温线图可以看出,在相对压力为P/P0=0.4~1之间,新鲜和使用后的催化剂均表现出具有回滞环的I型等温线。
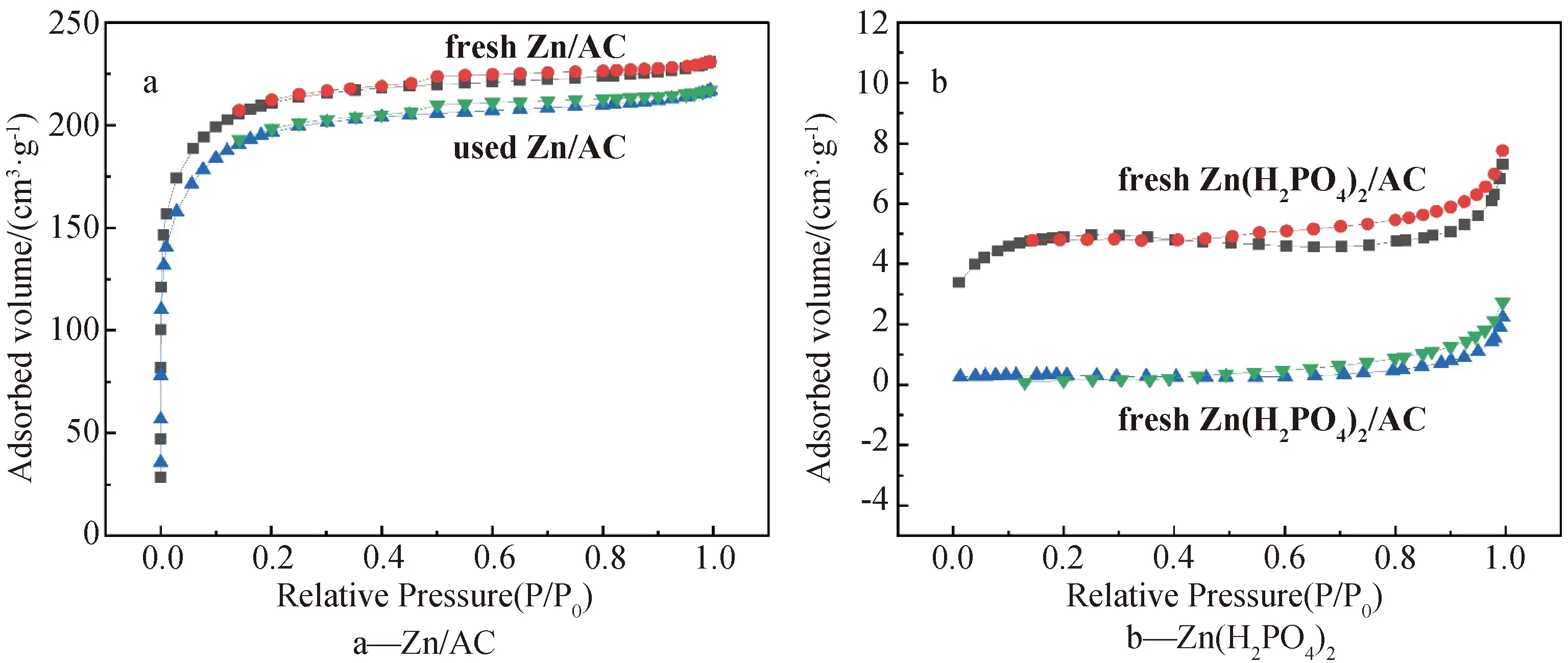
图2 AC催化剂的N2吸附脱附等温线图
同时表1显示,在反应前后Zn/AC的比表面积没有发生太大变化,这表明AC载体的结构并未遭到破坏。新鲜的Zn(H2PO4)2/AC催化剂的比表面积却很小,尤其是反应后,催化剂几乎没有了比表面积,表明AC载体的孔道结构可能遭到了破坏,一方面可能是由于磷酸的密度太大,制备的过程中全部附着在了AC载体表面,另一方面可能是制备过程中形成了大量的Zn(H2PO4)2占据了载体的孔道结构,进而导致催化剂比表面积以及孔容急剧减小。
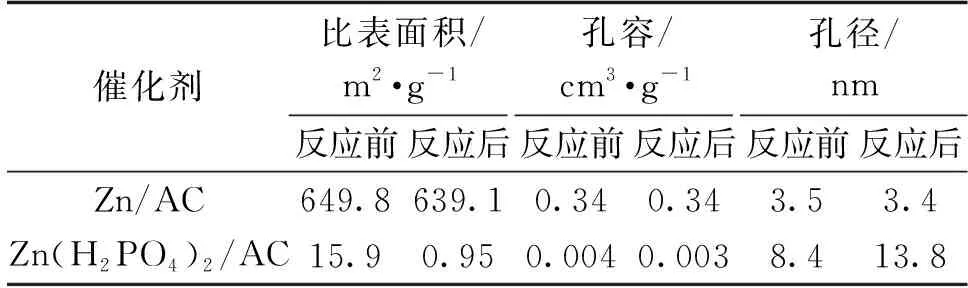
表1 催化剂的物理结构性质
2.3 XRD表征分析
从图3a显示的新鲜催化剂的XRD谱图可以看出,所有的样品在24.4°和43.7°处都出现了非晶态衍射峰,这可归因于活性碳的(002)和(101)晶面。Zn/AC催化剂除了AC载体的晶面特征峰外并未观察到其他衍射峰。新鲜的Zn(H2PO4)2/AC催化剂的谱图中发现了Zn(H2PO4)2·2H2O的衍射峰(PDF#27-0987),这表明Zn(H2PO4)2·2H2O成功负载至AC载体上,在23.8°处还出现了Zn2PO4(OH)的衍射峰。从图3b显示的反应后催化剂的XRD谱图能够看出,Zn(H2PO4)2/AC催化剂中除了Zn(H2PO4)2·2H2O和Zn2PO4(OH)的衍射峰外,还出现了Zn3(PO4)2·4H2O和ZnHPO4的衍射峰,这可能是由于在反应过程中Zn(H2PO4)2·2H2O发生了分解,BET分析结果中孔径的变化证明了这一点,同时这也可能是活性下降的原因。以上分析表明合成的Zn(H2PO4)2·2H2O成功进入AC载体孔道内,同时Zn/AC催化剂中载体结构保持完好,而Zn(H2PO4)2/AC催化剂中载体的孔道结构遭到了破坏,这与BET表征结果一致。
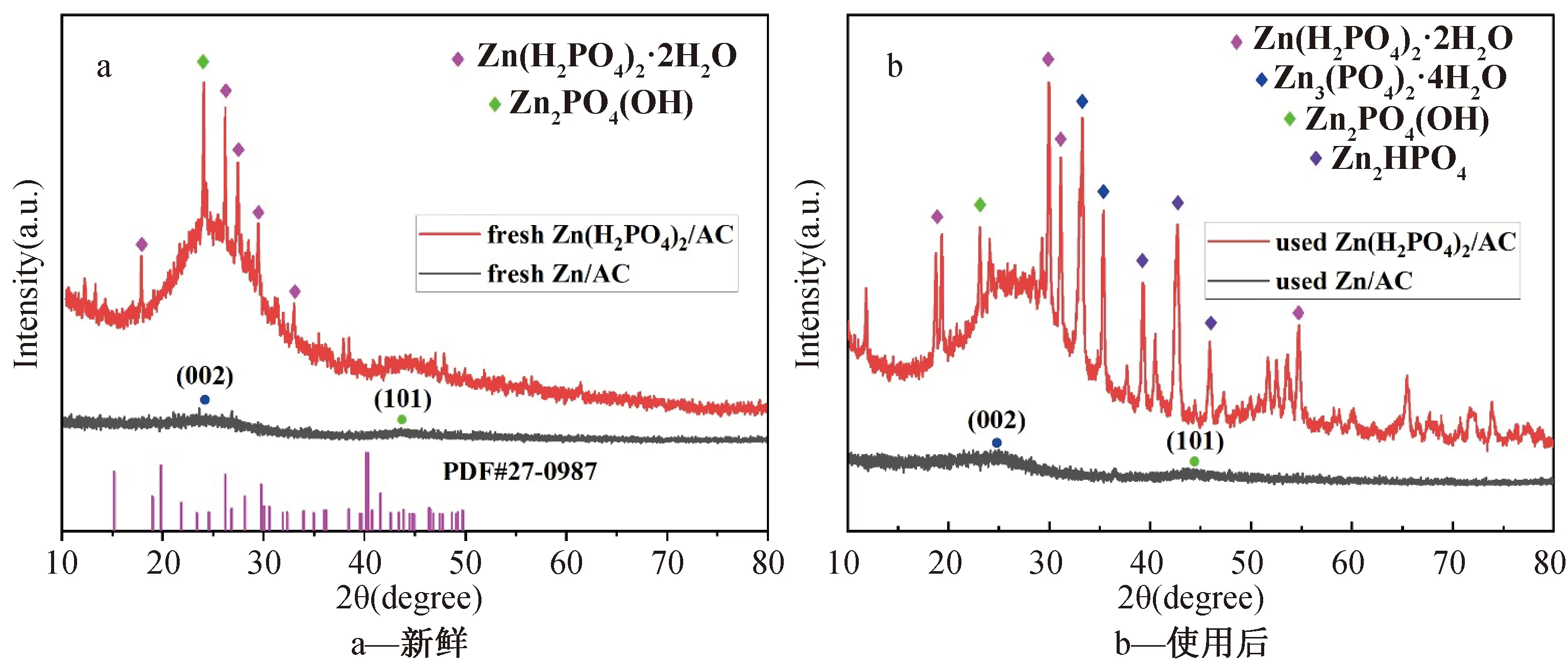
图3 催化剂的XRD谱图
2.4 Py-FTIR表征分析
研究表明,酸位点的存在虽然在一定程度上可以提高乙炔水合反应的活性,但它同时也是醇醛缩合反应的活性位点[15]。因此,为了进一步研究催化剂的酸位点及酸含量的变化,对新鲜催化剂进行了Py-FTIR表征分析,结果见图4和表2。根据文献,1 450 cm-1处的峰位代表Lewis酸位点,1 540 cm-1的峰位代表Bronsted酸位点,1 490 cm-1的峰位代表Lewis酸和Bronsted酸的结合位点[16-17]。如图4所示,催化剂在1 452 cm-1、1 492 cm-1、1 544 cm-1和1 611 cm-1的峰位分别对应于L酸位点、L+B酸位点、B酸位点和强L酸位点。此外,表2显示,Zn/AC催化剂得酸位点含量和总酸含量很低,Zn(H2PO4)2/AC催化剂中L酸位点含量、B酸位点含量以及酸含量均有大幅增加。相关研究表明,Zn2+能够与Bronsted酸位点结合形成(ZnOH)+物种[18]。因此,催化剂的酸位点和总酸含量的增加,有可能会促进Zn2+和Bronsted酸位点结合,这可能是Zn(H2PO4)2/AC催化剂性能更好,更稳定的原因。

表2 新鲜催化剂中酸位点的相对含量及总酸含量
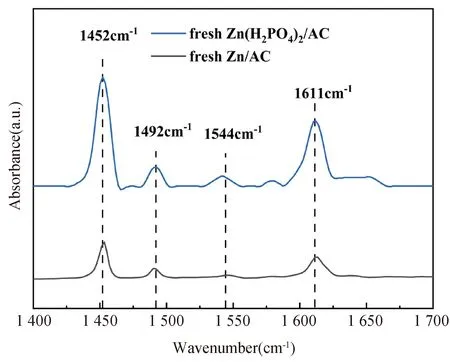
图4 新鲜催化剂的吡啶红外光谱
2.5 XPS表征分析
为了进一步了解Zn物种的化学状态及其含量,对新鲜和反应后的催化剂进行了XPS表征分析。从图5中可以发现,Zn/AC催化剂中Zn2p3/2的结合能为1 022 eV,相比之下,Zn(H2PO4)2/AC催化剂中Zn2p3/2的结合能向负方向发生偏移,这表明Zn得到了电子,周围的电子云密度增大,活性位点增多,这对反应是有利的,因此提高了催化剂的催化性能,这可能归因于磷酸基团的强配合能力。此外,从图5a和图5b中反应前后催化剂的XPS谱图中还可以看出,所有催化剂Zn2p3/2的XPS峰均可以拟合得到两个小峰,根据文献,位于高结合能的峰归属于(ZnOH)+物种,而低结合能的峰位归属于(ZnH)+物种[19]。从表3中可以看出,新鲜的Zn/AC催化剂中(ZnOH)+物种相对含量为34.6%,Zn(H)+物种占比65.4%,而Zn(H2PO4)2/AC催化剂中(ZnOH)+物种相对含量为59.9%,并且反应后催化剂中的(ZnOH)+物种略有减少,但仍保持在55%以上。(ZnOH)+物种含量略有减少,可能是(ZnOH)+在乙炔水合过程中的脱羟基化所致。两种催化剂的分析结果表明,更多的(ZnOH)+物种能够促进乙炔水合反应的进行,同时也证实了Py-FTIR表征分析中Zn2+能够与Bronsted酸位点结合形成(ZnOH)+物种的猜想。以上分析表明(ZnOH)+物种的增加,有利于反应的进行,提高乙醛的选择性。
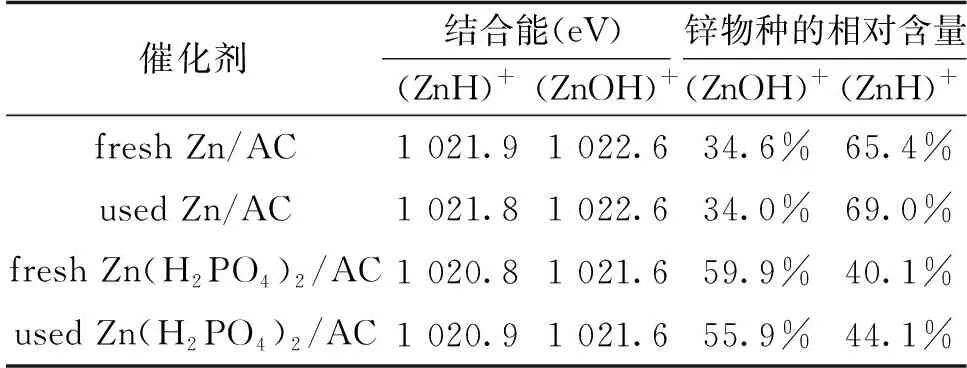
表3 催化剂中(ZnH)+和(ZnOH)+物种的相对含量
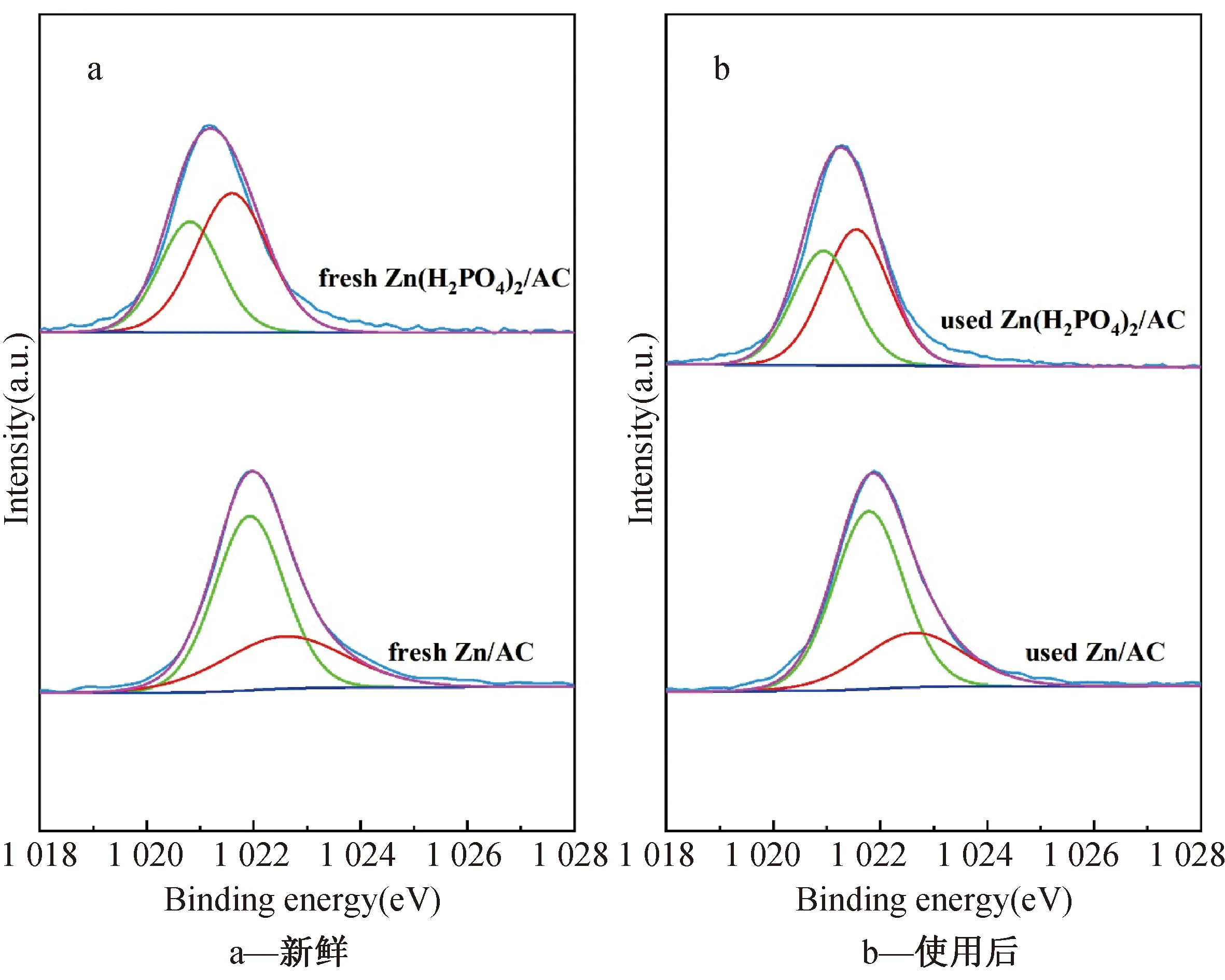
图5 催化剂的Zn2p3/2的XPS谱图
2.6 TG分析
研究表明,积碳是催化剂在反应过程中失活的一个重要原因,为了探究催化剂在反应过程中积碳的影响,本文将反应前后的催化剂在空气氛围下做了TG表征。图6展示了反应前后的不同催化剂在30 ℃~800 ℃区间内的失重情况。图中30 ℃~150 ℃的失重归因于催化剂中的结合水[20],这可能是与ZnCl2或Zn(H2PO4)2的易潮解性有关[21]。Zn/AC催化剂在350 ℃~550 ℃之间的失重归因于ZnCl2的分解。Zn(H2PO4)2/AC催化剂在150 ℃~350 ℃之间的失重,归因于磷酸或Zn(H2PO4)2的分解,而580 ℃~800 ℃时催化剂失重情况明显加深,这可能是由于磷酸或Zn(H2PO4)2的分解产物Zn3(PO4)2·4H2O和ZnHPO4进一步发生了分解或者载体表面发生了烧结。同时XRD表征结果中反应后的Zn(H2PO4)2/AC催化剂中Zn3(PO4)2·4H2O和ZnHPO4的衍射峰也证明了这一点。因此积碳量为反应前后催化剂在200 ℃~400 ℃阶段的失重量的差值。催化剂积碳量的计算结果如表4所示。从表4中可以发现,Zn/AC催化剂的积碳量为3.12%,而Zn(H2PO4)2/AC催化剂的积碳量为1.99%,表明后者的抗积碳性能更强。

表4 催化剂的积碳量
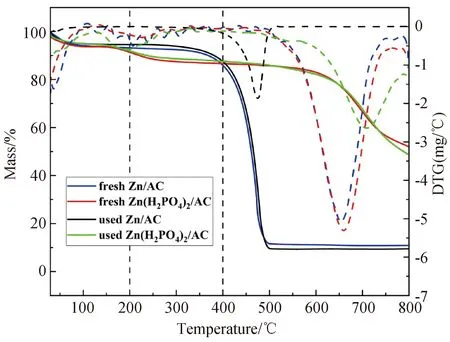
图6 反应前后不同Zn基催化剂的失重曲线
3 结论
(1)采用氧化锌和磷酸反应,利用过体积浸渍法合成了Zn(H2PO4)2/AC催化剂,反应10 h,乙炔的转化率由84.6%升至93.5%,而选择性也由75%降至68.6%,对比Zn/AC催化剂,反应活性得到明显的提升。
(2)BET和XRD结果表明,Zn(H2PO4)2成功负载至AC载体上,但在反应过程中部分分解成了Zn3(PO4)2·4H2O和ZnHPO4。
(3)Py-FTIR和TG结果表明:Zn(H2PO4)2/AC催化剂的酸位点含量以及总酸含量大幅增加,XPS分析证明了酸含量的增加促进了Zn2+与Bronsted酸位点结合,(ZnOH)+物种含量增多,同时积碳含量减少,从而促进了反应,提高了反应活性。
