结直肠癌样本免疫细胞浸润及肿瘤突变负荷分析
2023-06-08何仪佳韩聪玲王思睿轩小燕
王 娜,王 平,何仪佳,曾 晶,韩聪玲,王思睿,轩小燕,
(1.郑州大学基础医学院,河南 郑州 450001;2.黄河科技学院,河南 郑州 450006)
结直肠癌(colorectal cancer,CRC)是全球发病率第3、死亡率第4 的肿瘤,每年有超过120 万新增病例,60多万人死于CRC[1-3]。手术是目前治愈早期CRC的主要方法,但由于起病隐匿,多数患者发现时已有转移。化疗、放疗等延长了晚期CRC患者生存期,但治疗效果并不持久,耐药性不可避免地通过各种机制产生[4]。基因突变尤其是驱动基因如APC、TP53、KRAS的突变在不同肿瘤中突变频率不同,是影响肿瘤发生发展的重要因素[5-7],同时也影响肿瘤靶向治疗效果。以免疫检查点抑制剂尤其是PD-1/PD-L1 为代表的肿瘤免疫治疗是近年发展起来的非常有前景的治疗方法,在一些晚期CRC患者中具有显著疗效,但研究发现仅对微卫星不稳定性转移性CRC 患者有效[8],获益人群有限,仍急需开发新的治疗方法。研究CRC的肿瘤免疫细胞浸润微环境、基因突变图谱可为深入探讨CRC发生发展的分子机制以及为CRC的靶向治疗或免疫治疗提供理论基础。
转录组测序技术以及生物信息技术的发展,可以帮助我们多维度分析免疫细胞浸润、肿瘤细胞突变等影响肿瘤发生、发展、转移及治疗等重要因素。本文分析了来自癌症基因组图谱集(The Cancer Genome Atlas,TCGA)和基因表达综合数据库(Gene Expression Omnibus,GEO)的CRC 样本的免疫细胞浸润、基因集富集通路、肿瘤突变负荷等信息,为进一步研究CRC发生和转移的分子机制提供生物信息学数据支撑。
1 材料与方法
1.1 数据来源
从美国国家癌症研究所TCGA数据库(https://portal.gdc.cancer.gov/)下载473 例CRC 样本转录组表达数据、452 例CRC 患者临床信息数据以及399 例CRC 患者突变数据。从美国国家生物信息技术中心GEO 数据库(https://www.ncbi.nlm.nih.gov/geo/)下载GSE40967 数据集,含585 例CRC 患者样本芯片表达数据及临床信息,所有数据下载日期为2022年3月8 日。依据文献方法[9]将来自TCGA 数据库中的CRC 样本基因表达数据由FPKM 转化为TPM 后,与GSE40967 数据集的样本表达数据合并作为本文基因表达分析文件,采用ComBat 算法对数据进行批次校正。整理合并TCGA 及GEO 临床数据作为后续生存分析文件,在R4.2.0版本下运行数据。
1.2 CRC免疫细胞浸润分析
采用CIBERSORT 和ESTIMATE 算法分析CRC 样本基因表达数据,获得样本免疫评分、基质评分以及22种免疫细胞在样本中的比例[10]。采用Spearman检验分析样本中免疫细胞浸润程度、免疫评分和基质评分之间的相关性。根据CRC样本各免疫细胞比例的相似性,用ConsensusClusterPlus 将样本聚类为免疫细胞聚类A、B 两组,采用pheatmap 包绘制免疫细胞浸润热图。采用Kaplan-Meier 分析免疫细胞聚类A、B 两组间样本总生存期的差异。采用limma 包分析PD-L1基因表达在免疫细胞聚类A、B两组间的差异。
1.3 免疫细胞评分模型的构建及样本的生存分析和基因集富集分析
采用limma包分析免疫细胞聚类A、B两组间样本的所有差异表达基因(differentially expressed genes,DEG),以|Log2(FC)|>1,P<0.05 为过滤条件,FC 指基因表达的差异倍数(fold change)。采用Boruta 包查找DEG 的特征基因,用主成分分析法(principal component analysis,PCA)对特征基因降维分析获得每个CRC 样本的特征基因的特征值,采用类似基因表达指数的方法[11]利用公式“免疫细胞评分=∑PC1A-∑PC1B”(即每个样本的所有A 组特征基因的特征值减去所有B 组特征基因的特征值)建立CRC 样本的免疫细胞评分模型,对每个样本进行评分。采用surv_cutpoint 函数查找评分的最佳临界值,根据最佳临界值将CRC样本分为免疫细胞评分高、低两组。采用Kaplan-Meier 分析组间样本总生存期差异。对样本进行基因集富集分析(gene set enrichment analysis,GSEA),阐明免疫细胞评分高、低两组样本的基因富集通路。
1.4 肿瘤突变负荷计算及生存分析
肿瘤突变负荷(tumor mutation burden,TMB)指每百万碱基中发生置换、插入/缺失突变的总数,用来评估肿瘤的基因突变频率。从TCGA 下载CRC 突变数据,统计非同义突变数目,计算样本TMB。用surv_cutpoint 函数查找TMB 最佳临界值,根据最佳临界值将CRC样本分成肿瘤突变负荷高、低两组,采用Kaplan-Meier 分析组间样本总生存期差异,采用Maftool绘制样本基因突变瀑布图。
1.5 统计分析
采用Wilcoxon 检验进行两组之间比较,Kaplan-Meier(log-rank 检验)进行生存分析,Spearman 进行相关性分析。P<0.05为差异具有统计学意义。
2 结 果
2.1 CRC样本免疫细胞浸润分析
免疫细胞浸润分析发现,CRC 肿瘤组织内CD8+T细胞和初始记忆性CD4+T 细胞(r=-0.34,P<0.05)、活化的记忆性CD4+T 细胞(r=0.37,P<0.05)、M0 巨噬细胞(r=-0.47,P<0.05)具有相关性。中性粒细胞和活化的浆细胞具相关性(r=0.41,P<0.05)。在22种免疫细胞中,免疫评分仅与M1 巨噬细胞呈弱相关(r=0.32),而与其他21 种细胞均无明显相关(|r|<0.3,P>0.05,图1A)。根据CRC 样本中浸润的免疫细胞比例的相似性,采用ConsensusClusterPlus 对样本进行聚类为免疫细胞聚类A和B两群。CD8+T细胞、M0巨噬细胞、活化的记忆性CD4+T细胞等呈现明显聚类(图1B)。生存分析提示免疫细胞聚类A、B 两组患者总生存期无显著差异(P=0.928,图1C)。PD-L1基因表达水平在两组之间差异无统计学意义(P>0.05,图1D)。
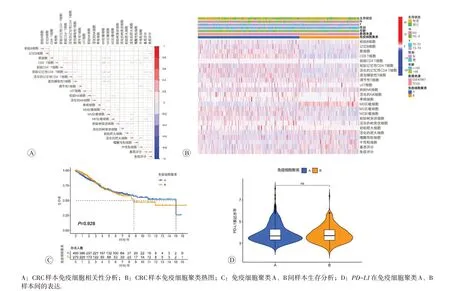
图1 CRC样本免疫细胞浸润分析
2.2 CRC 样本免疫细胞评分模型建立及基因集富集分析
采用limma包分析免疫细胞聚类A、B组间样本的DEG,采用Boruta 包查找DEG 的特征基因,依据文献方法将特征基因分为A、B 两组,若特征基因的相对表达量在免疫细胞聚类A组样本呈高表达而在免疫细胞聚类B 组样本低表达,该基因定义为A 组特征基因,反之定义为B 组特征基因[12]。经计算,共获得28个特征基因,其中A 组特征基因18 个,分别为CLCA1、IGHM、IGLJ3、ZG16、ITLN1、ADH1C、CLCA4、CA2、CA4、PIGR、UGT2B17、MS4A12、HEPACAM2、FCGBP、PLA2G2A、CEACAM7、SPINK4、REG4;B 组特征基因10 个,分别为MMP9、SPP1、COL10A1、THBS2、COL11A1、FNDC1、CXCL5、COMP、SFRP4、CXCL8。依据CRC 样本免疫细胞评分模型,计算每个CRC 样本的免疫细胞评分,采用surv_cutpoint 函数查找最佳临界值,依据临界值将CRC样本分为免疫细胞评分高、低两组。生存分析提示免疫细胞评分高组的患者总生存期长于免疫细胞评分低组的患者(P=0.005,图2A)。
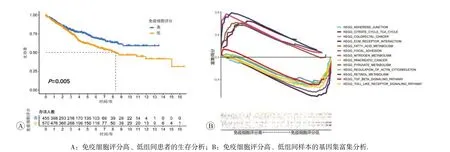
图2 CRC样本生存分析及基因集富集分析
GSEA 分析显示,免疫细胞评分高组的基因主要富集在KEGG_FATTY_ACID_METABOLISM,NITROGEN_METABOLISM、KEGG_CITRATE_CYCLE_TCA_CYCLE、KEGG_PYRUVATE_METABOLISM、KEGG_RETINOL_METABOLISM 等信号通路,这些通路在碳水化合物、脂肪酸代谢中发挥重要作用,与肠道的正常生理功能密切相关。值得注意的是,未见免疫相关基因在免疫细胞评分高组样本中富集。免疫细胞评分低组的基因主要富集在KEGG_COLORECTAL_CANCER,ECM_RECEPTOR_INTERACTION, KEGG_REGULATION_OF_ACTIN_CYTOSKELETON 等信号通路,这些通路与癌症的形成、发展和转移密切相关,这也跟免疫细胞评分低组患者的低生存期相一致(图2B)。
2.3 肿瘤突变负荷分析
TMB 是肿瘤对免疫治疗反应的潜在生物标志物[13],较高的TMB增加了肿瘤新抗原产生的概率。本文分析了免疫细胞评分高、低两组样本的基因突变数据,用maftool绘制突变瀑布图,列出变异频率最高的前20个基因。值得注意的是,两组之间突变频率最高的20个基因一致,这一结果和其他课题组分析的结果一致[14-16]。CRC 肿瘤驱动突变基因APC突变频率在免疫细胞评分高、低组分别为77%和73%,TP53分别为47%和57%,KRAS分别为45%和41%。APC基因以移码插入突变和移码缺失突变为主,TP53、KRAS基因以错义突变为主(图3A、3B)。TMB 在免疫细胞评分高、低组间的差异无统计学意义(P=0.17,图3C)。生存分析提示肿瘤突变负荷低组患者总生存期长于肿瘤突变负荷高组患者(P=0.035,图3D)。
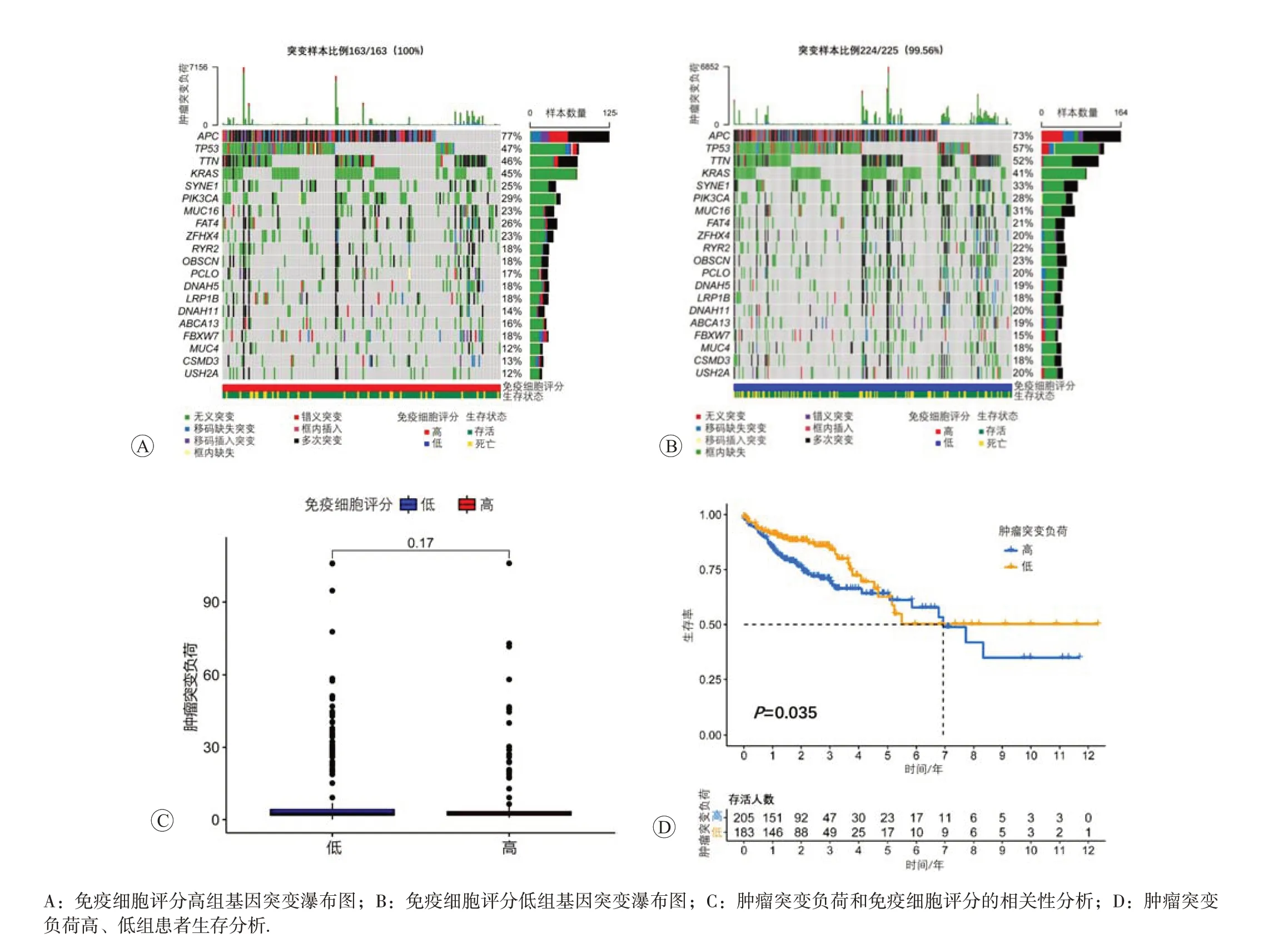
图3 CRC肿瘤突变负荷分析
3 讨论
CRC是常见的恶性肿瘤,手术、放疗、化疗以及免疫治疗在一定程度上延长了肿瘤患者的生存时间,但目前的治疗方法均不理想。深入分析肿瘤的免疫细胞浸润、肿瘤突变负荷等信息,可为研究影响CRC发生发展的关键因素以及为CRC的治疗提供理论基础。
为了降低不同测序平台对结果的影响,本研究将来自TCGA 数据库的CRC 样本转录组数据如前所述标准化后和来自GEO 数据库的GSE40967 数据集进行合并。通过对免疫细胞浸润分析,未发现在CRC样本中具有强相关性的免疫细胞,免疫细胞聚类A、B 两群患者之间总生存期差异无统计学意义,这些结果提示在CRC 样本中抗肿瘤免疫未充分活化发挥抗肿瘤作用。PD-L1的表达是免疫检查点抑制剂是否有效的主要指标[17],本文分析发现PD-L1表达水平在免疫细胞聚类A、B 两组之间差异无统计学意义,这也解释了临床观察到的大多数CRC患者对免疫检查点抑制剂靶向治疗无反应。
为进一步探索影响CRC 患者生存差异的关键因素,本文构建了CRC样本免疫细胞评分模型,依据免疫细胞评分将样本分成免疫细胞评分高、低两组。免疫细胞评分高组患者总生存期长于免疫细胞评分低组患者,提示该模型具有潜在预测CRC 患者预后的价值。GSEA 分析发现脂肪酸代谢、氮代谢、碳代谢等主要的物质代谢信号通路在免疫细胞评分高组富集,提示免疫细胞评分高组样本的活跃基因以参与生理代谢为主,推测免疫细胞评分高组样本肿瘤细胞分化程度高,执行生理功能相关基因具有表达优势。有趣的是,一些跟肿瘤发生、发展密切相关的通路在免疫细胞评分低组样本中富集,提示免疫细胞评分低组样本分化程度低,恶性程度高,这也跟免疫细胞评分低组患者的低生存率相一致。据此,我们推测免疫细胞评分高、低组患者之间的生存差异主要由与肿瘤发生发展相关的一些信号通路决定。GSEA 分析发现在免疫细胞高、低组均未见有免疫活化相关通路富集,这也和上述免疫细胞浸润观察到的结果一致,提示在CRC样本中,肿瘤免疫应答未充分活化而发挥抗肿瘤作用。
肿瘤的发生由基因突变积累引起,这些基因所编码的蛋白质往往是细胞周期信号通路中的重要蛋白质,控制细胞分裂和生长的驱动基因发生突变,在肿瘤的发生、发展、转移、免疫逃逸以及治疗耐药中发挥关键作用[18-19]。CRC 的基因组已经进行了广泛深入的研究[20-21],APC、KRAS、TP53等基因为CRC 的驱动基因[22],在CRC 发生发展和转移中发挥重要作用。APC突变导致APC/β-catenin 通路的过度活化[23-24],KRAS突变使其下游信号通路异常[25],抑癌基因TP53突变后产生的蛋白导致其生理功能丧失,这些驱动基因以不同的方式参与细胞增殖、侵袭和转移[26-27]。TMB 分析发现APC、KRAS、TP53的突变在免疫细胞评分高、低组样本中均占有较大的比例,结合上述免疫细胞评分高、低组GSEA 分析结果,我们认为,在CRC 样本中,与机体产生的抗肿瘤免疫作用相比较,驱动基因的突变是决定肿瘤恶性程度以及肿瘤发展和转移的主要因素。本文数据为进一步深入研究CRC发生和转移机制提供了必要的生信数据支撑。
