吲唑酮并噌啉类化合物的合成及结构表征
2023-06-07宋嘉霖陈望梁姚林声远林鑫成舒兵
宋嘉霖,陈望梁,姚林声远,林鑫成,舒兵
(1.广东药科大学新药研发中心,广东 广州 510006;2.广东药科大学药学院,广东 广州 510006)
吲唑酮衍生物是重要的含氮杂环化合物,存在于一系列的药物分子和功能材料中[1-3]。一些稠合的和官能团化的吲唑酮衍生物(代表性化合物的结构式见图1)具有多种良好的生物活性,如抗炎、抗肿瘤、抗生育、抗高血压等,已经引起了广泛关注和研究[4-6]。吲唑酮类化合物作为吡咯酮的生物电子等排体,在药物化学和各类药物研究中较常见[7]。其中,氨基侧链和嘧啶基取代的吲唑酮类化合物在抗癌活性方面表现出潜在活性而备受关注[8-9]。此外,吲唑酮衍生物具有优异的光学性能,可以用作有机半导体和荧光探针[10-12]。因此,开发有效和实用的合成稠合吲唑酮衍生物的方法具有非常重要的意义。

图1 含有稠合吲唑酮骨架的代表性生物活性化合物Figure 1 Representative bioactive compounds containing fused indazolone skeleton
目前,合成稠合吲唑酮的方法已有不少文献报道[13-15]。据文献报道,吲唑酮可以作为导向基团,在金属Rh(Ⅲ)、Ir(Ⅰ)、Pd(Ⅱ)的催化下以串联反应的方式合成稠合吲唑酮杂环(图2a)。例如,Perumal等[16]报道了N-苯基吲唑酮为导向基团与炔烃的活化反应,该反应通过Rh(Ⅲ)催化C-H/N-H [4+2]环化,用于合成吲唑并[1,2-a]噌啉。2018 年,Sakhuja课题组使用α-重氮羰基化合物作为C-C 键合成子,在Ir(Ⅰ)催化下与1-芳基吲唑酮发生[4+2]环化反应,合成了稠合的吲唑酮[17]。该课题组于2021 年和2022年,先后报道了1-芳基吲唑酮通过Rh(Ⅲ)催化的C-H 活化/烯烃插入/还原与各种硝基烯烃的反应[18]和1-芳基吲唑酮在Pd(Ⅱ)催化下与丙烯酸进行C-H 活化/[4+2]环化反应构建稠合的吲唑酮[19]。此后,Hu等[20]开发了一种使用碘鎓叶立德作为环化试剂合成稠合吲唑酮骨架的方法。尽管上述方法为合成稠合吲唑酮类化合物提供了很好的策略,但是对过渡金属催化吲唑酮的C-H 官能化和进一步环化反应的探索,以及通过新反应模式和新偶联试剂构建稠合吲唑酮化合物的方面来看,仍具有很大的研究空间。

图2 吲唑酮导向的过渡金属催化的C-H活化/环化反应Figure 2 Indazolone directed transition metal catalyzed C-H activation/annulation reactions
本研究拟采用过渡金属催化剂三价铑,通过1-苯基吲唑酮和碳酸亚乙烯酯之间的C-H/N-H 环化反应,建立反应条件温和、原料易于获得、操作简单的合成稠合吲唑酮类衍生物的新方法(图2b)。
1 实验部分
1.1 仪器与试剂
DF-101S 集热式恒温加热磁力搅拌器(河南巩义予华有限公司);ZF-7A 手提式紫外分析仪(杭州齐威仪器有限公司);N-1300V-W 旋转蒸发仪(上海艾朗仪器有限公司);ME104 电子天平、SHB-III 循环水式多用真空泵(巩义市科瑞仪器有限公司);Advance Bruker 400M 超导核磁共振波谱仪(瑞士BRUKER 公司),以CDCl3或DMSO-d6为溶剂;柱层析硅胶(200~300 目硅胶)和G254薄层色谱硅胶(山西诺泰生物科技有限公司);[Cp*RhCl2]2、碳酸亚乙烯酯(分析纯,上海毕得医药有限公司),其余所有溶剂如DCE、HFIP、DCM、EA 等均为市售分析纯,用前未经处理,直接使用。
1.2 合成方法
将0.10 mmol 1-苯基吲唑酮(1)、0.12 mmol碳酸亚乙烯酯(2)、0.002 5 mmol [Cp*RhCl2]2,0.010 mmol AgSbF6、0.10 mmol CsOAc、0.50 mL 六氟异丙醇(HFIP)依次加入15 mL 的耐压瓶中,旋紧塞子,在磁力搅拌下于60 ℃连续反应12 h,薄层色谱(TLC)检测反应过程。反应结束后,用旋转蒸发仪减压除去溶剂,所得粗产物用硅胶柱层析分离[二氯甲烷-甲醇(体积比80∶1~40∶1)],即得到目标产物3。合成路线见图3。
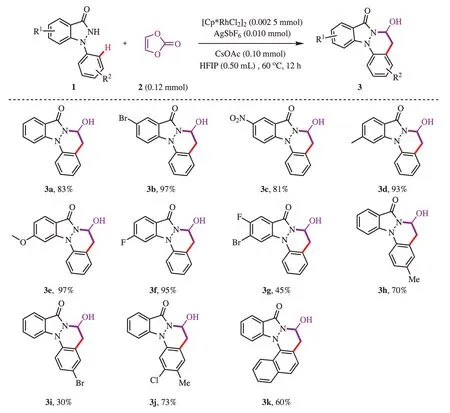
图3 1-苯基吲唑酮类化合物的合成Figure 3 Synthesis of 1-arylindazolone compounds
1.3 克级实验
将5.0 mmol 6-甲基-1-苯基吲唑酮(1d)、6.0 mmol碳酸亚乙烯酯(2)、0.125 mmol[Cp*RhCl2]2,0.50 mmol AgSbF6、5.0 mmol CsOAc、25 mL 六氟异丙醇(HFIP)依次加入100 mL 的耐压瓶中,旋紧塞子,在磁力搅拌下于60 ℃连续反应12 h,薄层色谱(TLC)检测反应过程。反应结束后,用旋转蒸发仪减压除去溶剂,所得粗产物用硅胶柱层析分离[二氯甲烷-甲醇(体积比80∶1~40∶1)],即可得到目标产物3d。
1.4 控制实验
将0.10 mmol 1-苯基吲唑酮(1a)、0.12 mmol 碳酸亚乙烯酯(2)、0.002 5 mmol [Cp*RhCl2]2或0.010 mmol AgSbF6、0.10 mmol CsOAc、0.50 mL 六氟异丙醇(HFIP)依次加入15 mL 的耐压瓶中,旋紧塞子,在磁力搅拌下于60 ℃连续反应12 h,薄层色谱(TLC)检测反应过程。
2 结果与讨论
2.1 化合物的鉴定
6-羟基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3a):淡黄色固体。1H NMR(400 MHz,DMSO)δ: 7.98(d,J=8.6 Hz, 1H), 7.84 (d,J=7.7 Hz, 1H), 7.78 (d,J=8.1 Hz, 1H), 7.70 (dd,J=11.5, 4.1 Hz, 1H), 7.38(dd,J=13.3, 7.2 Hz, 2H), 7.22 (t,J=7.4 Hz, 1H),7.12 (t,J=7.4 Hz, 1H), 6.84 (d,J=3.2 Hz, 1H), 6.31(d,J=1.6 Hz, 1H), 3.21-3.05 (m, 2H)。13C NMR(101 MHz, DMSO)δ:156.58, 139.02, 135.04, 133.06,130.51, 127.68, 123.84, 123.32, 122.43, 121.24, 116.06,114.26, 111.11, 70.11, 34.48。 HRMS (ESI)m/zcalcd. for C15H12N2O2Na [M+Na]+275.0797; Found 275.0802。
10-溴-6-羟基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3b):淡黄色固体。1H NMR(400 MHz,DMSO)δ:7.96(d,J=9.0 Hz,2H),7.84-7.71(m,2H),7.39(t,J=7.7 Hz,2H),7.15(t,J=7.4 Hz,1H),6.94(d,J=3.9 Hz,1H),6.37-6.27(m,1H),3.21-3.14(m,1H),3.14-3.07(m,1H)。13C NMR(101 MHz,DMSO)δ:155.58,137.91,135.86,134.92,130.96,128.12,126.37,124.14,123.03,118.15,114.91,113.67,113.20,70.69,34.76。 HRMS(ESI)m/zcalcd.for C15H11BrN2O2Na[M+Na]+352.990 2;Found 352.991 0。
6-羟基-10-硝基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3c):黄色固体。1H NMR (400 MHz, DMSO)δ:8.61 (s, 1H), 8.44 (d,J=7.5 Hz, 1H), 8.13 (d,J=9.2 Hz, 1H), 7.87 (d,J=7.7 Hz, 1H), 7.45 (d,J=7.1 Hz,2H), 7.27 (d,J=7.2 Hz, 1H), 7.06 (d,J=2.5 Hz, 1H),6.42-6.28 (m, 1H), 3.25-3.11 (m, 2H)。13C NMR(101 MHz, DMSO)δ: 156.26, 141.29, 140.36, 133.91,131.14,128.27,128.22,125.56,124.21,121.13,116.02,115.85, 111.83, 71.02, 34.67。HRMS (ESI)m/zcalcd.for C15H11N3O4Na[M+Na]+320.064 8;Found 320.065 1。
6-羟基-11-甲基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3d):黄色固体。1H NMR (400 MHz, DMSO)δ:7.81(d,J=8.0 Hz,2H),7.71(d,J=8.0 Hz,1H),7.45-7.32 (m, 2H), 7.17-7.09 (m, 1H), 7.04 (d,J=8.0 Hz,1H), 6.81 (d,J=3.7 Hz, 1H), 6.34-6.25 (m, 1H),3.20-3.03 (m, 2H), 2.50 (s, 3H)。13C NMR (101 MHz, DMSO)δ: 157.07, 144.05, 139.99, 135.54,130.87, 128.02, 123.85, 123.54, 123.26, 122.77,114.73, 114.31, 111.17, 70.45, 34.94, 22.41。HRMS(ESI)m/zcalcd. for C16H14N2O2Na [M+Na]+289.095 3;Found 289.095 9。
6-羟基-11-甲氧基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3e):黄色固体。1H NMR(400 MHz,DMSO)δ: 7.81 (d,J=8.2 Hz, 1H), 7.71 (d,J=8.7 Hz, 1H),7.45-7.34 (m, 2H), 7.30 (s, 1H), 7.13 (t,J=7.4 Hz,1H), 6.81 (dd,J=8.7, 1.5 Hz, 1H), 6.77 (d,J=3.9 Hz,1H), 6.29-6.22 (m, 1H), 3.93 (s, 3H), 3.19-3.04 (m,2H)。13C NMR (101 MHz,DMSO)δ:164.06,157.10,141.09,135.44,130.86,128.13,125.33,123.74,123.15,114.91, 111.31, 109.83, 94.18, 70.55, 56.27, 35.04。HRMS (ESI)m/zcalcd. for C16H14N2O3Na [M+Na]+305.090 2;Found 305.090 5。
11-氟-6-羟基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3f):淡黄色固体。1H NMR (400 MHz, DMSO)δ:7.94-7.72 (m, 3H), 7.46-7.30 (m, 2H), 7.21-7.10 (m,1H), 7.09-6.99 (m, 1H), 6.87 (d,J=8.8 Hz, 1H),6.36-6.23 (m, 1H), 3.23-3.02 (m, 2H)。13C NMR(101 MHz,DMSO)δ:165.90 (d,J=247.5 Hz),156.48,139.84(d,J=12.4 Hz),134.92,130.89,128.15,126.59(d,J=10.4 Hz), 124.10, 122.94, 115.02, 113.11,110.15 (d,J=24.8 Hz), 98.29 (d,J=28.9 Hz), 70.58,34.82。19F NMR (376 MHz, CDCl3)δ: -105.49。HRMS (ESI)m/zcalcd. for C15H11FN2O2Na [M+Na]+293.070 3;Found 293.070 8。
11-溴-10-氟-6-羟基-5H-吲唑并[1,2-a]噌啉-8(6H) - 酮(3g): 黄色固体。1H NMR (400 MHz,DMSO)δ: 8.34 (d,J=5.1 Hz, 1H), 7.83 (d,J=7.4 Hz, 2H), 7.45-7.35 (m, 2H), 7.16 (t,J=7.4 Hz, 1H),6.96 (d,J=3.9 Hz, 1H), 6.36-6.25 (m, 1H), 3.17 (d,J=15.5 Hz, 1H), 3.09 (d,J=15.3 Hz, 1H)。13C NMR(101 MHz, DMSO)δ:155.85 (d,J=3.9 Hz), 153.64(d,J=239.7 Hz), 136.17, 134.87, 130.87, 128.21,124.26, 122.96, 116.38 (d,J=7.6 Hz), 115.98, 115.37(d,J=24.0 Hz),115.09,110.45(d,J=25.0 Hz),70.90,34.72。19F NMR (376 MHz, CDCl3)δ:-116.52。HRMS (ESI)m/zcalcd. for C15H10BrFN2O2Na [M+Na]+370.980 8;Found 370.981 0。
6-羟基-3-甲基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3h):白色固体。1H NMR (400 MHz, CDCl3)δ: 7.79(d,J=7.9 Hz,1H),7.60(d,J=8.5 Hz,1H),7.51(t,J=7.7 Hz, 1H), 7.41 (d,J=8.0 Hz, 1H), 7.14-7.03 (m,3H), 6.39-6.32 (m, 1H), 3.14 (dd,J=15.1, 3.1 Hz,1H), 3.04 (dd,J=15.1, 2.9 Hz, 1H), 2.29 (s, 3H)。13C NMR (101 MHz, CDCl3)δ: 157.81, 138.77, 134.14,132.81, 132.56, 131.02, 128.36, 124.25, 122.29,121.16, 115.54, 114.16, 110.51, 71.52, 34.25, 20.85。HRMS (ESI)m/zcalcd. for C16H14N2O2Na [M+Na]+289.095 3;Found 289.095 8。
3-溴-6-羟基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3i):白色固体。1H NMR (400 MHz, DMSO)δ:7.96(d,J=8.5 Hz, 1H), 7.85 (d,J=7.7 Hz, 1H), 7.75 (d,J=8.7 Hz, 1H), 7.71 (t,J=7.7 Hz, 1H), 7.61 (s, 1H),7.54 (dd,J=8.7, 2.2 Hz, 1H), 7.25 (t,J=7.5 Hz, 1H),6.87(d,J=4.1 Hz,1H),6.30(d,J=3.3 Hz,1H),3.21-3.07(m,2H)。13C NMR(101 MHz,DMSO)δ:157.02,139.36,134.71,133.57,133.27,130.63,125.50,124.30,121.99, 116.68, 116.52, 115.14, 111.59, 70.30, 34.64。HRMS (ESI)m/zcalcd. for C15H11BrN2O2Na [M+Na]+352.990 2;Found 352.990 8。
2-氯-6-羟基-3-甲基-5H-吲唑并[1,2-a]噌啉-8(6H)-酮(3j):黄色固体。1H NMR(400 MHz,CDCl3)δ:7.66(d,J=8.0 Hz,1H),7.53-7.44(m,2H),7.42(s,1H),7.21-7.14(m,2H),7.03(t,J=6.9 Hz,1H),6.46-6.42 (m, 1H), 3.14 (dd,J=15.3, 2.2 Hz, 1H), 3.00(dd,J=15.2, 3.1 Hz, 1H), 2.32 (s, 3H)。13C NMR(101 MHz, CDCl3)δ:157.72, 138.65, 133.57, 133.21,132.93,132.56,131.65,124.43,121.44,120.65,115.85,114.59, 110.36, 71.45, 33.75, 19.49。HRMS (ESI)m/zcalcd. for C16H13ClN2O2Na [M+Na]+323.056 4;Found 323.056 4。
8-羟基-7H-苯并[h]吲唑并[1,2-a]噌啉-10(8H)-酮(3k):白色固体。1H NMR (400 MHz, DMSO)δ:8.06(dd,J=6.0,3.4 Hz,1H),7.93-7.85(m,2H),7.82(dd,J=6.1, 3.3 Hz, 1H), 7.66-7.60 (m, 2H), 7.59-7.54 (m, 1H), 7.53 (d,J=8.2 Hz, 1H), 7.19 (t,J=7.4 Hz, 1H), 6.98 (d,J=8.5 Hz, 1H), 6.85 (d,J=3.4 Hz,1H), 6.42-6.36 (m, 1H), 3.19 (dd,J=15.5, 1.9 Hz,1H), 3.10 (dd,J=15.5, 2.1 Hz, 1H)。13C NMR (101 MHz,DMSO)δ:159.54,142.70,133.93,132.97,130.63,129.14,128.32,126.49,126.43,126.17,125.26,124.86,124.28, 123.94, 121.27, 116.23, 111.52, 73.05, 36.11。HRMS (ESI)m/zcalcd. for C19H14N2O2Na [M+Na]+325.095 3;Found 325.096 0。
2.2 反应条件的优化
以1-苯基吲唑酮(1a)和碳酸亚乙烯酯(2)为模板反应,先后考察了催化剂、反应温度、添加剂、银盐、溶剂等反应条件对产物3a 收率的影响,结果见表1。可见:不同的金属催化剂中,以[Cp*RhCl2]2的催化效果最佳;适当升高反应温度会使产物收率增加,但是温度过高则会导致产率降低,所以在60 ℃的条件下有利于反应的进行;添加剂[B(OH)3、PivOH、Na2CO3、KH2PO4、CsOAc]对反应结果也产生明显的影响,其中CsOAc 可以有效促进反应的发生;对反应中银盐进行筛选的结果表明,AgSbF6是最适合的银盐;当使用的溶剂由DCE 替换为HFIP时,反应收率提高到了83%。

表1 反应条件的优化Table 1 Reaction condition optimization
综上所述,最佳反应条件为:1-苯基吲唑酮(0.1 mmol),碳酸亚乙烯酯(0.12 mmol),催化剂[Cp*RhCl2]2(0.002 5 mmol),银盐AgSbF6(0.01 mmol),添加剂CsOAc(0.1 mmol),反应溶剂六氟异丙醇(HFIP,0.5 mL),60 ℃下反应12 h。
2.3 目标化合物的合成
在得到了最佳的反应条件后,考察该反应条件的底物适应性。结果表明,含有各种不同官能团的1-苯基吲唑酮(1)都可以转化为目标产物3。当化合物1 的C5 或C6 位置为单取代基时,获得目标产物的产率较高,尤其是C5 位置溴取代,C6 位置甲基、甲氧基、氟取代时,产率高达97%(3b、3d、3e、3f),C5 位置是硝基取代时,也依然有81%的产率(3c)。但是C5、C6 位置为二取代基的1-苯基吲唑酮就表现出了较弱的相容性(3g)。当1-苯基位置有取代时,显现出相对较低的反应产率。1-苯基4位为甲基时,产率较高,达70%,但1-苯基4 位溴取代时则表现出了较低的产率,只有30%(3h、3i、3j)。这可能说明1-苯基上的取代基情况对该反应产率影响较大。此外,在最优条件下,1-萘基取代的吲唑酮也依然耐受,得到了相应的产物(3k)。
通过以上底物扩展结果推测:当吸电子基和给电子基在化合物1 的C5 或C6 位置单取代时,对反应的影响不大;但当1-苯基上为吸电子基单取代时,对反应的影响较大,可能是由于1-苯基上的吸电子基影响了C-H活化过程,导致反应产率较低。
2.4 克级实验
通过上述实验,发现该反应条件的底物适应性良好,为进一步使该催化方法被推广及应用,在标准条件下进行了克级实验。最终以88%的产率得到所需的吲唑酮衍生物3d(图4)。在最优条件下,将6-甲基-1-苯基吲唑酮1d(5.0 mmol,1.12 g)与碳酸亚乙烯酯2(6.0 mmol,516.3 mg)反应12 h,最终得到产物1.17 g,产率88%。

图4 克级实验Figure 4 Gram-scale experiment
2.5 控制实验
为了深入了解该合成反应的机制,进行了相应的控制实验(图5)。结果显示,该反应在不添加催化剂[Cp*RhCl2]2或银盐AgSbF6的情况下均未检测到目标产物3a的生成,表明铑催化剂和银盐是该反应中不可缺少的要素。
2.6 可能的反应机制
根据初步的机制研究数据和以往的文献,本文提出了一种合理的反应机制(图6):[Cp*RhCl2]2、AgSbF6和添加剂CsOAc 之间进行配体交换生成了活性催化剂Cp*Rh(Ⅲ)物种Ⅰ,Cp*Rh(Ⅲ)物种Ⅰ与底物1a 通过协同的C-H 活化过程形成关键的铑配合物Ⅱ,随后与碳酸亚乙烯酯配位并发生迁移插入得到铑配合物Ⅲ;铑配合物Ⅲ的Rh-C键发生迁移且同时脱去一分子CO2后生成中间体Ⅳ,再经过质子化反应得到中间体Ⅴ,并且使Cp*Rh(Ⅲ)催化剂再生,用于下一个催化循环;最后中间体Ⅴ发生分子内环化反应,得到最终产物3a。

图6 可能的反应机制Figure 6 Mechanistic proposal
3 结论
建立了一种以1-苯基吲唑酮和碳酸亚乙烯酯为原料,[Cp*RhCl2]2作为催化剂,六氟异丙醇作为反应溶剂,通过三价铑催化的C-H/N-H 环化反应生成稠合吲唑酮并噌啉类衍生物的合成方法。该反应具有条件温和、原料易于获得、产率高(97%)、对官能团兼容性好等优点,为具有吲唑酮骨架的结构复杂多样活性化合物或药物分子的合成提供了一条新型的合成路径。
