抗N-甲基-D-天冬氨酸受体脑炎合并髓鞘少突胶质细胞糖蛋白抗体相关疾病一例
2023-06-06陈伟曹菁菁于翠玉刘卫国
陈伟 曹菁菁 于翠玉 刘卫国
1 病例报告患者男,39岁。因“左眼视物不清1年余,言行异常、口齿不清5 d”于2021-1-26入院。2019年11月出现左眼视物模糊,伴视野缺损、视物白茫,遂就诊当地医院,完善颅脑MRI检查提示左侧胼胝体白质病变,脱髓鞘可能。行抗髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体及血清抗水通道蛋白-4(AQP4)抗体检测均阴性。诊断“脱髓鞘性脑病”。给予激素治疗(具体不详)后症状完全消失出院。5 d前患者无明显诱因出现傻笑,第2天晨起出现言语不清,重复言语,大喊大叫等,持续约1~2 h,并有四肢发作性抖动,1 d内发作多次。遂就诊于作者医院急诊,以“自身免疫性脑炎?”收入院。既往史无特殊,否认有家族遗传病史、家族肿瘤病史。入院查体:内科查体无明显异常;意识清楚,精神差,记忆力、定向力及计算力检查不能合作;双侧瞳孔等大等圆,直径3 mm,直接及间接对光反射灵敏,各向眼动充分,眼震(-),视力视野检查不合作;双侧额纹对称,悬雍垂居中,咽反射存在,伸舌居中;四肢肌力5级,肌张力正常;双下肢腱反射活跃,病理征阴性;指鼻试验及跟膝胫试验不配合,脑膜刺激征阴性。血常规检查:白细胞计数 10.16×109/L,中性粒细胞76.0%。脑脊液(CSF)检查:压力20 mmH2O(1 mmH2O=9.8 Pa),免疫球蛋白G及白蛋白正常,葡萄糖5.57 mmol/L,蛋白0.46 g/L,有核细胞计数81×106个/L,CSF自身免疫性脑炎12项检测结果显示抗N-甲基-D-天冬氨酸受体(NMDAR)抗体1∶100,中枢脱髓鞘6项检测显示抗髓鞘少突胶质细胞糖蛋白(MOG)抗体1∶10。血清自身免疫性脑炎12项检测结果显示抗NMDAR抗体1∶100。甲型、乙型、丙型、丁型、戊型肝炎病毒(甲型、乙型、丙型、丁型、戊型)、HIV、梅毒检测结果正常,肾功能、甲状腺功能、肿瘤标记物以及尿便常规检测结果正常。头颅MRI平扫及增强扫描(2019-12-27)可见左侧胼胝体片状长T1、长T2信号,T1增强扫描后未见异常强化灶,扩散加权成像扫描未见异常信号(图1)。2021-1-28复查头颅MRI平扫未见明显异常(图2)。结合临床表现、影像学特征、血清及脑脊液相关抗体检查结果,诊断为抗NMDAR脑炎合并MOG抗体相关疾病(MOG-IgG associated disorders,MOGAD)。给予患者免疫球蛋白静脉滴注〔0.4 mg/(kg·d),×5 d〕和甲强龙冲击治疗。甲强龙起始剂量500 mg,每6 d减半,减至120 mg时维持6 d,改为口服醋酸泼尼松片(60 mg,1次/d)并联合口服吗替麦考酚酯(早晚各0.5 g)治疗。给予口服奥卡西平(300 mg,2次/d)控制肌阵挛发作,奥氮平控制精神症状。患者精神症状大约在治疗2周左右改善,同时肌阵挛症状逐步消失。经28 d治疗患者出院,出院时患者交流顺畅,无异常行为,认知功能正常,肢体活动无障碍。出院后6个月随访,患者未再出现精神行为异常,无肌阵挛发作。复查抗NMDAR抗体水平为1∶10。
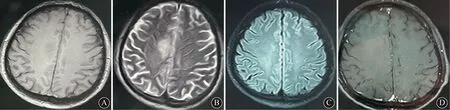
A:T1像显示左侧胼胝体片状长T1信号;B:T2像显示左侧胼胝体片状长T2信号;C:扩散加权成像扫描未见异常信号;D:T1增强扫描未见异常强化灶

注:A:T1像;B:T2像
2 讨论自身免疫性脑炎是一类由自身免疫机制介导的脑炎,抗NMDAR抗体脑炎是其中最常见类型,主要症状表现为精神行为异常、记忆障碍、癫痫发作等。目前研究认为自身免疫性脑炎多为单一抗体致病,该例患者抗NMDAR抗体和MOG抗体均阳性。MOG是中枢神经系统炎性脱髓鞘疾病中重要的自身免疫抗体和细胞免疫反应的靶点,近年来学者将MOGAD作为一种独立疾病谱[1],患者常见临床表现为一侧或双侧起病的视神经炎,常可累及眼眶结缔组织,其他症状包括脊髓炎、急性播散性脑脊髓膜炎、脑干脑炎等。该例患者初次发病以单侧视神经炎为主要表现,并伴有脑白质脱髓鞘表现,对免疫治疗反应良好。患者首次发病时MOG抗体检测阴性,第2次入院后复查MOG抗体阳性,推测患者初次发病很可能与MOG抗体相关。目前有关MOGAD与抗NMDAR脑炎协同发病的机制尚不完全清楚。有研究显示,自身免疫性脑炎患者在免疫抑制剂减量甚至中止免疫治疗时,其自身免疫系统在修复过程中可能诱导多种致病抗体[2]。该例患者即是在结束第一次免疫治疗后不久再次出现神经系统受损症状。多抗体叠加阳性的自身免疫性脑炎患者临床表现较为复杂,既可以同时出现不同抗体致病的特征表现,也可以在相当长的时间内先后发病。Rojc等[3]报道的1例抗NMDAR脑炎合并MOGAD患者两种症状相隔长达13年。该例患者出现抗NMDAR脑炎症状的时间与出现视神经炎的间隔长达14个月。
综上所述,临床中抗NMDAR脑炎合并MOGAD的患者虽较少见,但两者可能存在共同的免疫致病机制,应注意扩大可疑患者的抗体筛查范围。此类患者接受免疫治疗后大多数预后良好,但较单一抗体致病的自身免疫性脑炎需适当延长免疫治疗时间,以预防病程复发。
