计算机辅助药物设计在天然抗肿瘤药物研究中的应用
2023-06-03徐度玲李鸿岩柳佳娣
徐度玲, 李鸿岩,6*, 张 红,6, 柳佳娣
(1.中国科学院近代物理研究所 医学物理研究室,甘肃 兰州 730000; 2.中国科学院重离子束辐射生物医学重点实验室,甘肃 兰州 730000; 3.甘肃省重离子辐射医学应用基础研究重点实验室,甘肃 兰州 730000;4.中国科学院大学 核科学与技术学院,北京 100039; 5.先进能源科学与技术广东省实验室,广东 惠州 516006;6.甘肃省同位素实验室,甘肃 兰州 730300)
人类在漫长的医学探索过程中,逐渐积累了利用天然药物治疗疾病的经验.在中医理论指导下,扶正固本复方制剂仍用于食管癌、胃癌等气阴两虚兼热毒证患者在放化疗时的合并用药.随着提取技术的不断发展,学者尝试从天然药物中分离有效成分,开创了天然药物应用的新纪元,并衍生出天然产物这一概念,即从动物、植物和微生物中分离出的生物二次代谢产物.1803年,法国药剂师Derosne从罂粟属植物鸦片中分离出那可丁;1805年,德国药物学家Sertürner从鸦片中分离得到吗啡;1853年,法国化学家查尔斯·盖哈特从柳树皮中分离出乙酰水杨酸,开启了天然产物提取和分离的先河.紫杉醇又名红豆杉醇,是从红豆杉属树皮中提取的天然次生代谢化合物,适用于治疗卵巢癌和乳腺癌,是美国食品和药物管理局批准的第一个天然抗癌药物[1—2],但它对外周神经的毒性限制了在临床上的应用.恶性肿瘤是人类死亡的主要因素,开发安全有效、作用机制明确及毒副作用小的天然产物,成为癌症治疗药物研发工作的重中之重.计算机辅助药物设计(computer-aided drug design,CADD)的兴起,为天然产物的开发提供了强大支持.CADD可避免盲目实验导致的资金浪费,做到有的放矢,并缩短药物的研发周期.笔者基于上述研究背景,综述近年来CADD技术在天然抗肿瘤药物研发中的应用,以期为抗肿瘤药物的研发提供参考.
1 CADD在天然产物开发中的作用
癌症的治疗药物最初是单一药物,一般只涉及单一靶点.由于恶性肿瘤的形成原因复杂,单一药物治疗时易产生耐药性且导致严重的副作用[3].大量的研究表明,大部分药物能够作用于多个靶点,但具有多个有效成分的天然产物的作用机制复杂.因此通过结构修饰来增大药效、降低毒性和耐药性是学者关注的主要问题.通过生物信息学和计算方法预测天然产物的作用靶点,既可减少科研资源的浪费,又可避免前期开发天然产物投入巨大资金但终止于各期临床试验阶段的情况发生[4].根据类药性对已知天然产物的结构和性质进行统计分析,归纳出天然产物成为新药应具有的特性.例如,辉瑞公司资深药物化学家Lipinski等对比小分子化合物与成药化合物的结构,得到口服小分子化合物的化学特性,并归纳为“五规则”,该规则有助于研究人员快速初筛出潜在药物[4—5].以药物动力学的吸收(absorbtion,A)、分布(distribution,D)、代谢(metabolism,M)、排泄(excretion,E)和毒性(toxic,T)即ADMET为基础建立数学模型,可分析药物在体内的时间-浓度变化关系.通过预测药物的动力学性质和毒性,可有效避免药物间的拮抗作用.药物代谢动力学数据库有助于研究人员快速判断天然产物的毒性.随着生物大分子和小分子信息的迅速增加,CADD通过预测药物靶点、改造药物结构和预测药物毒性等减少了科研资源的浪费,提高了药物筛选的成功率,为天然产物的开发和应用提供了技术保证.CADD是用于化合物存储、管理、分析和建模的计算工具和来源,包括用于设计化合物的计算机程序、系统评估潜在药物的工具以及用于开发化学结构相互作用的数据存储库[6].根据常用药物发现的计算方法,将CADD技术分为基于结构的药物设计(structure-based drug design,SBDD)、基于配体的药物设计(ligand-based drug design,LBDD)和基于片段的药物发现(fragment-based drug discovery,FBDD).
2 基于结构的药物设计(SBDD)
SBDD方法适用于已通过实验方法或者理论模拟预测方法得到的受体或受体-配体复合物的三维结构体系[7],通常是蛋白质或核酸,可确定与大分子生物功能相关的关键位点及其相互作用.SBDD主要包括从头设计(denovo)和虚拟筛选(vritual screening,VS).SBDD过程中往往也通过分子动力学(molecular dynamics,MD)模拟,深入了解配体与靶蛋白的结合模式,研究相互作用过程中的结构变化[8].
2.1 基于结构的靶标预测
靶标的识别与预测是现代药物研发的第一步.所谓靶标,是药物作用于生命体中的生物大分子,如蛋白质、核酸及其复合物.基于已知的靶点设计和筛选新型药物,是抗肿瘤药物研发的第一步,且筛选靶点的成功率直接关系药物的前期试验和临床试验.许多疾病由多个靶点作用导致,癌症的致病因素更复杂,因此,研发多靶点抗肿瘤药物是肿瘤治疗药物研发的重要策略.天然产物及其衍生物的多靶点特性,为多靶点抗肿瘤药物的研发提供了参考.通过CADD技术预测药物-靶点的相互作用,有助于识别潜在配对的“药物-靶点”,加速天然抗肿瘤药物的研发和应用进程.
根据靶标的三维结构,表征药物和靶标的结合位点,建立蛋白质靶标库,并通过打分函数对潜在的靶标按匹配度进行筛选[9],从而寻找自由能最低以及配体与受体结合的最优构象.这种筛选方法包括反向分子对接和反向药效团匹配.反向分子对接方法是在给定一个化合物结构的情况下,分析该分子与一个或多个靶标蛋白质的互补作用,再根据结合的紧密程度进行排序筛选.反向分子对接方法已广泛用于药物和天然化合物的新靶点筛选、解析药理机制以及判断药物的毒副作用等研究.陈宇综于2001年提出反向分子对接概念[10],并开发了第一个反向对接程序INVDOCK.2006年,蒋华良开发了一种新的靶标垂钓算法——TarFisDock[11].林荣信于2012年开发了一种反向找靶的免费在线服务系统——IDTarget[12].靶标预测工具的相继出现进一步拓宽了反向分子对接技术的应用范围.
天然三萜类化合物的结构具有多样性,对许多疾病的治疗显现出积极作用,如优异的药理活性[13].积雪草酸(asiatic acid,AA)(图1)是从积雪草(伞形科植物)中提取的具有乌苏烷骨架的五环三萜类化合物,可用于缓解疼痛和咳嗽[14].1971年,学者发现积雪草酸可用于治疗皮肤创伤[15].随后对积雪草酸的其他药理功效进行广泛的研究,研究结果显示,积雪草酸具有多种药理学特征,包括抗炎、抗菌[16]、抗氧化、心肌保护、神经保护[17]和抗肿瘤[18]等.研究人员最初发现,积雪草酸通过凋亡的级联反应抑制细胞增殖和促进细胞死亡[19],进而发挥对人胆管癌的抗肿瘤活性[20],且积雪草酸对正常组织的毒性小[21].此外,积雪草酸能有效抑制肺癌[22]、皮肤癌[23]、卵巢癌[24]、舌癌[25]、肾细胞癌[26]和乳腺癌[27]等癌细胞的增殖,具有广谱抗癌的特性.但因积雪草酸的药理作用弱、水溶性差及生物利用度低等缺点,限制了其在临床上的应用[28].近年来,研究人员通过对积雪草酸的结构进行改造,如对A环的结构进行修饰(羟基修饰、缩环修饰、扩环修饰和开环修饰)、C环的结构进行修饰(C-28酯化和酰胺化修饰)和其他修饰[28],研发出大量的积雪草酸衍生物,且这些衍生物表现出良好的抗肿瘤效果.因此,林碧琪等使用CADD方法,以积雪草酸为先导化合物,对积雪草酸的结构进行修饰[29],以血管内皮生长因子(vascular endothelial growth factor,VEGF)为靶点,合成2类(C-28位成酯和C-28位成酰胺)共10个具有抗肿瘤活性的衍生物.结果表明,化合物11(图2)和化合物13(图3)通过氢键或疏水键与靶蛋白周围的关键氨基酸ASN75,GLU42,GLU73和LEU97相互连接.后续通过细胞毒活性实验比较积雪草酸的目标衍生物、吉非替尼(gefitinib)和阿霉素(adriamycin)对人胃癌细胞(SGC7901)和肺癌细胞(A549)的IC50.结果显示,阿霉素的抑制效果最好,化合物11和化合物13的抑制效果均优于吉非替尼和母体积雪草酸,但C-28位成酰胺化合物13的抑制效果优于C-28位成酯化合物的.
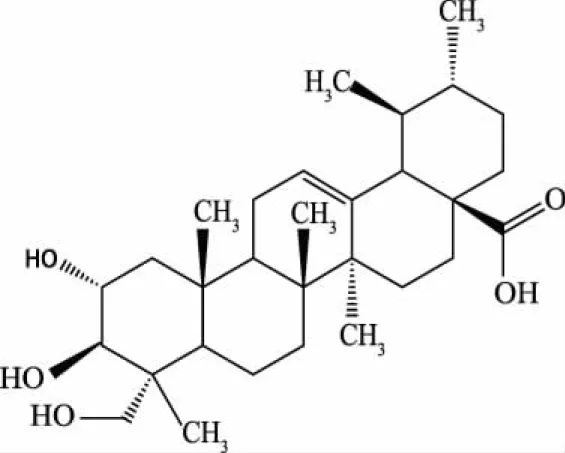
图1 积雪草酸的化学结构
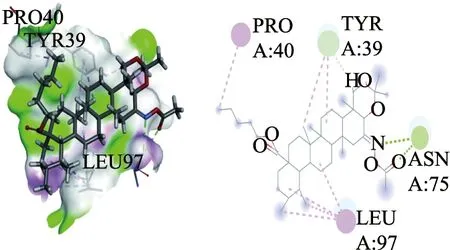
图2 化合物11与VEGF的结合位点[29]

图3 化合物13与VEGF的结合位点[29]
在松树精油中存在着单烯萜家族中最具代表性的一种天然化合物——α-蒎烯(α-pinene)(图4).研究表明,α-蒎烯通过不同的分子机制表现出抗癌活性.Zhao等通过PC-3细胞系在裸鼠中建立皮下异种移植肿瘤模型[30],发现α-蒎烯能显著抑制人类前列腺癌细胞的增殖,并诱导细胞凋亡和细胞周期阻滞.与未经α-蒎烯处理的对照组小鼠相比,α-蒎烯能够抑制肿瘤的发展.Kusuhara等将小鼠置于含α-蒎烯的环境中[31],发现在富含α-蒎烯环境中,小鼠的黑色素瘤细胞生长被抑制.然而,α-蒎烯具有结构不稳定性、亲和力差及活性不足等缺点[32],使α-蒎烯的应用受到限制.杨梦蝶等以α-蒎烯结构为基础[33],在保留萜类优势结构的基础上进行氧化、还原及酯化修饰,分别得到两种α-蒎烯衍生物,即(1R,5S)-(6,6-二甲基二环[3,1,1]庚-2-烯-2-基)苯磺酸酯和(1R,5S)-(6,6-二甲基二环[3,1,1]庚-2-烯-2-基)对甲苯磺酸酯.通过AutoDock 4.2,以衍生物为配体、肿瘤细胞周期阻滞和凋亡相关通路因子为受体,建立对接体系,发现合成的两种α-蒎烯衍生物能够与细胞周期蛋白依赖性激酶2(cyclin dependent kinase 2,CDK2)稳定结合,推测衍生物可能导致肿瘤细胞发生S期阻滞.对比氨基酸的类型和对接能,发现化合物A(图5)较化合物B(图6)与CDK2的结合更紧密.细胞实验也证实这两种衍生物能够抑制肿瘤细胞增殖[32, 34].
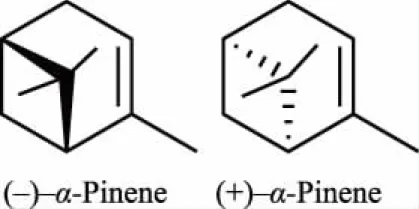
图4 α-蒎烯的化学结构
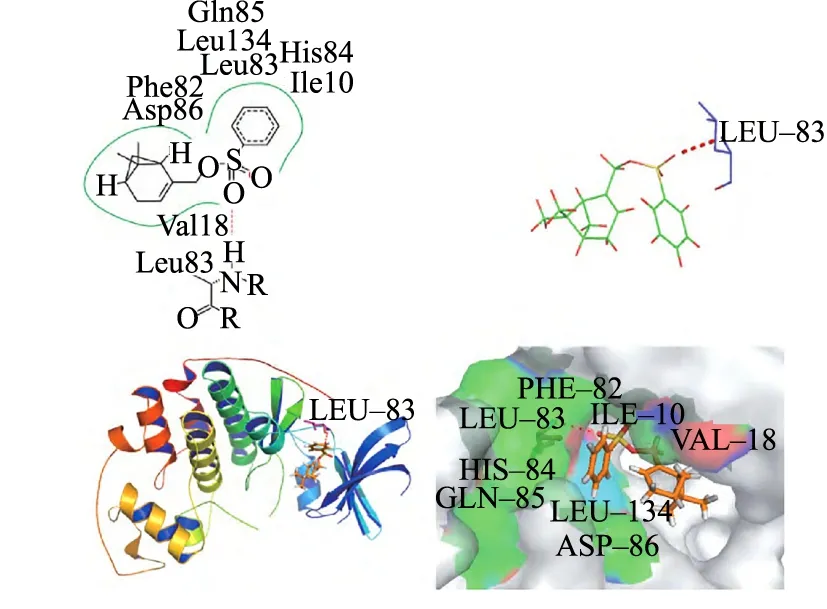
图5 化合物A与CDK2的结合模式[33]
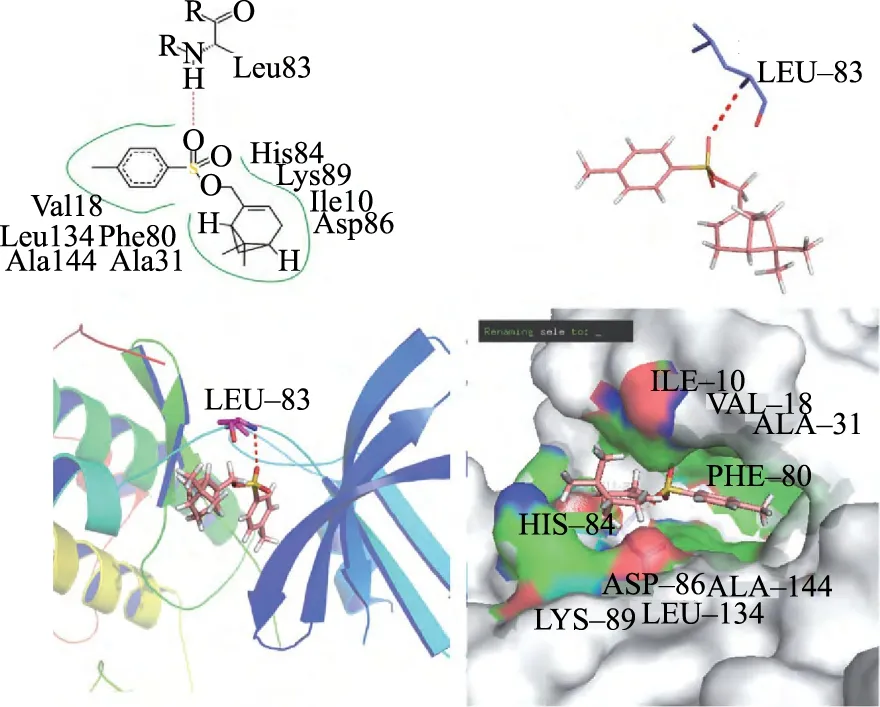
图6 化合物B与CDK2的结合模式[33]
杜国华等以灵芝子实体中的26种三萜类化合物为配体[35],通过Discovery Studio对11种靶点蛋白进行反向对接,结果显示,有8种灵芝三萜类化合物具有抗肿瘤活性,其中5个具有多靶点作用;细胞实验进一步证实,灵芝酸Y具有开发成抗肺癌药物的潜力,7-oxo-ganoderic acid Z2和ganoderon B也可作为抗肿瘤药物研发的候选化合物.
随着分析技术的革新,单一的反向对接模式也暴露出一些问题:目标蛋白的结合位点易错、小分子数据库筛选不当、对接模式的选择和高对接分数与分子动力学模拟不符以及化合物是抑制剂还是激动剂的界限不清楚等.为了解决上述问题,反向药效团匹配技术随之产生并逐步成熟.反向药效团匹配是将给定的化合物与多个药效团匹配,根据匹配程度确定潜在靶标.李洪林等建立在线预测服务系统PhamMapper,该系统是目前最大的药效团数据库[36].Inte:Ligand公司通过药效团生成工具Ligand-Scout[37],开发了三维药效团数据库.Yuan等通过基于药物的逆向扫描平台PharmMapper以及药物靶点数据库[38],对加密素的抗癌靶点进行评估和快速预测.首先,基于京都基因与基因组百科全书(Kytot Encyclppedia of Genes and Genomes,KEGG)通路数据库筛查抗癌靶点的采集路径.随后,预测隐性酮病影响的潜在药理活性和通路,验证使用全细胞测试筛选的目标.结果显示,共筛选出8个具有抗癌潜力的靶点:MAP2K1,RARα,RXRα,PDK1,CHK1,AR,Ang-1R,Kif11.这些靶点主要与癌症发展的4大因素有关,包括细胞周期、血管生成、细胞凋亡和雄激素受体.
2.2 虚拟筛选
虚拟筛选指在进行生物活性筛选前,根据预先设定的条件,在计算机上对化合物分子进行预筛选,识别出最可能与靶标结合的小分子,从而大大降低需要进行实验测试的化合物种类,提高先导化合物的发现效率.虚拟筛选以虚拟或者实体的化合物数据库为对象,对预处理化合物进行初筛,是对传统实验的有效补充,大大提高了实验筛选的效率,降低了筛选成本.虚拟筛选技术于1997年提出[39].根据靶标三维结构是否已知,虚拟筛选又可分为基于靶标结构和配体结构的虚拟筛选,当两者信息都具备时将两种方法结合使用,可大大提高虚拟筛选的效率[40].
基于结构的虚拟筛选,首先需要获得靶标的三维结构,其结构可以是晶体结构、溶液结构或者构建的模型结构.无配体的结构以及不同配体的复合物结构,具有不同的分辨率,因此需对这些结构进行分析,选择合适的结构进行筛选.对选定的靶标结构进行预处理,补齐晶体结构中缺失的原子和残基;补充晶体结构中缺失的氢原子,并分配需要的电荷;确定活性口袋带电残基的质子化状态;去除对配对没有影响的水分子等.将小分子三维结构数据库与靶标活性口袋结合,寻找两者最佳的作用模式及其结合构象.由于分子对接时面临呈现相反状态的速度和精度的选择,一般先进行多轮筛选,然后再进行合理的打分与生物活性测试.
在虚拟筛选过程中常用到分子对接方法.对接一般指两个或多个分子通过几何匹配和能量匹配原则相互识别的过程.分子对接方法的最初思想来源于酶和底物的“锁钥”模式[41].1982年,Kuntz提出分子对接,并开发了第一个分子对接软件Dock[42].分子对接方法的流程:确定靶标的结构,将配体与靶标结合,寻找靶标的结合口袋,分析两者之间的位置、取向和构象等变化,使用打分函数对比结合模式.打分函数大致可分为[43]基于力场、经验、知识、描述符以及一致性等类型.
Wang等以G蛋白偶联受体15(G protein-coupled receptor 15,GPR15)为受体[44],从含有62 500个小分子的数据库中,首先通过SYBYL中的Surflex-Dock模块,根据结合口袋的形状和静电势的相似性,进行基于形状的虚拟筛选,获得733种化合物;然后缩小范围,重新对接以获得与GPR15结合的最优构象;最后根据设定的阈值(13.00,8.00)评分,按最低结合能挑选出8个GPR15的潜在拮抗剂(图7).
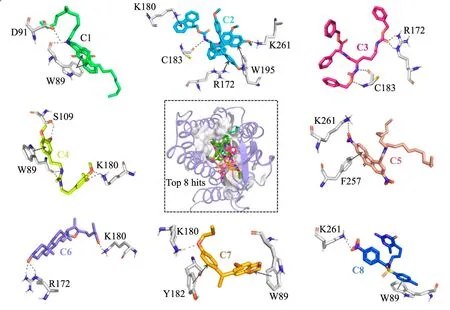
注:紫色线代表氢键,黑线表示pi-pi和pi-阳离子相互作用.
苍术酮是苍术的重要成分之一,具有抗肿瘤作用,但其作用靶点目前尚不明确.胡翠英等通过Autodock进行分子对接[45],模拟分析结果表明,苍术酮与AKT,MMP-9和Bcl-xl等蛋白的功能区域存在相互结合作用; AKT,MMP-9和Bcl-xl可能是苍术酮的潜在作用靶点.这在一定程度上补充了苍术酮的抗肿瘤机理,同时为以苍术酮为先导化合物开发新型抗肿瘤靶向药物的研究提供了参考.董江红等以薯蓣皂苷元为基础[46],通过AutoDock 4.2对接、设计薯蓣皂苷元衍生物,并选择性地合成12种薯蓣皂苷元衍生物.在人恶性黑色素瘤细胞A375、人肺腺癌细胞A549、人肝癌细胞HepG-2和人慢性髓原白血病细胞K562肿瘤细胞中,进行体外抗肿瘤活性实验,对人肾上皮细胞H293和人肝细胞LO2进行毒性实验,结果表明,新合成出的12种化合物大多具有良好的抗肿瘤活性,且对H293,LO2正常细胞无毒或低毒.目前,分子对接过程中仍存在一些问题,例如,在匹配相互识别过程中,受体大分子通常被假设为刚性.由于蛋白质分子和配体小分子均发生构象变化,很难同时考虑两者的柔性势能[47].分子对接可简化为刚性对接、柔性对接和半柔性对接.刚性对接的前题是研究体系的构象不发生变化,如软对接方法;柔性对接主要应用于精确考查分子的直接识别情况,研究体系的构象变化.如苏博源参考PharmaDB药效模型团数据库,对凹叶木兰、中缅木莲、紫玉兰中所提取的37种化合物,按照全柔性分子对接的筛选条件[48],分析每种配体与结合性最好的受体之间的相互作用机制.从通过反向分子对接筛选得到的33种化合物中,选出对接稳定的23种化合物,做后续ADMET测试,寻找具有成药性的化合物.半柔性对接方法主要应用于处理药物小分子与蛋白质之间的对接,且配体的构象允许在一定范围内变化,如Kuntz等开发的分子对接方法[49].
DOCK程序已广泛应用于药物分子设计领域[50].此外,Trott等开发的AutoDock Vina[51],能够实现快速计算,同时显著提高了绑定模式预测的准确性.2014年,Chen等通过PharmMapper和idTarget服务器[52]、 PyRx 0.8中的分子对接程序AutoDock Vina进行实验,得出丹参素的潜在抗癌靶标可能是HRas的结论.
3 基于配体的药物设计
3.1 基于配体的靶标预测
基于配体的靶标预测是,比较分析需要预测靶标的化合物和具有已知靶标活性的分子结构,主要使用相似性搜索和机器学习方法,其中相似性搜索是基于配体的靶标预测最简单且最可靠的方法之一[53].二维结构相似性主要通过分子指纹(如PubChem)描述,再通过计算比较两个分子的相似性,如Keiser等开发的Similarity Ensemble Approach(SEA)软件[54—55].Dunkel等基于二维结构相似性的药物分类和靶标预测开发了在线SuperPred软件[56].三维结构相似性是建立活性配体三维构象库、计算要预测的化合物与构象库中活性分子三维结构的匹配程度,从而预测未知化合物的靶标.根据Kimelford和Wahba的理论[57],机器学习的实质也基于相似性,不同点在于它们对选取的特征进行进一步的抽象分析,并根据抽象分析的相似性重构数据的组织形式.Sharma等提出基于机器学习的DTI预测计算框架[58],该框架成功地对不同靶点的药物进行排序,通过降维和主动学习方法,处理类别间不平衡数据,从而实现对药物与靶点间的相互作用进行预测.近年来,研究人员使用多种手段进行靶标预测.Huang等提出“most-similar ligand-based target inference” ( MOST)[53],它是通过查询并利用化合物最相似配体的指纹相似性和显性生物活性进行靶标预测的方法,具有准确度高和稳定性好的特点.同时,通过对氟安松和芦荟大黄素进行分析,证明MOST与FDR控制程序的集成方法,这就提供了一个可以预测已知药物新靶点和天然产物中活性成分作用机制的可靠框架.
Zhang等提出命名为Evis的提高虚拟筛选准确度的方法[59],该方法基于配体-受体的结合模式,对筛选出的化合物中前1%进行排名,并使用3D构象方法进行配体相似度计算,且进行DUDE,LIT-PCBA和DEKOIS 2.0数据集基准测试.该方法通过局部评估并结合配合物口袋-配体结构相似度计算,改善分子对接虚拟筛选的性能,可有效避免过度拟合问题.
3.2 药效团建模
药效团通常指可以与受体结合位点形成氢键、静电、范德华力作用或疏水作用的原子和官能团及其相互之间的空间关系.药效团的提出最早可追溯到1909年.德国科学家Ehrlich在染料研究中提出结合基团和毒性基团,并认为这些基团对生物活性有重要影响[60].后来,研究者提出许多对于药效团的解释,呈现出“百花争鸣”的局面.1998年,国际纯粹与应用化学联合会(IUPAC)提出药效团的定义:与特定的靶标结合并引发(或阻断)靶标生物响应所必需的空间和电子特征集合[61].其中,空间和电子特征作为抽象特征不包括具体官能团的描述.该定义与最初的定义不相符.为统一描述,有学者提出药效团是分子中决定生物效应的特征模式.因此,当不同分子的药效团模型相同时,它们具有相同的生物靶标结合位点,表示它们具有相似的生物学效应特征[62].
药效团模型的构建首先需要分析和确定配体含有的药效团特征,其特征一般用球体表现,即以球心为准确位置,以半径为可偏离的范围[63],具体特征包括氢键供体(受体)、正(负)电中心、疏水中心、芳环中心和排除体积等.天然产物通常在“桥头”有更多的sp3杂化原子,且有更多的手性中心.一个完整的药效团模型除包括上述特征外,还需满足一定的空间限制,如药效团特征之间的距离和角度等.
药效团模型通常基于一组配体生成,即将配体叠加,从中选出生物活性分子所必需的相互作用特征:生物活性小分子的选取—小分子生物三维结构的生成和构象分析—药效团模型的构建—模型评价及验证—数据库检索验证.在挑选合适的生物活性小分子时,一般搜集到的小分子只有二维结构,须将二维结构转化为三维结构.在构建模型时,要对小分子进行构象分析.对于构建好的药效团模型,根据小分子的结构特征和匹配程度进行分析,选取最优的模型并进行多次验证,最后通过数据库进行搜索,发现新的药物先导化合物.
4 药物代谢动力学与毒性预测
在新药研发中,造成药物研发失败的主要原因:缺乏药效、体内毒性、药物代谢动力学性质差、副作用明显等.药物代谢动力学(pharmacokinetics,PK)是研究体内药物浓度随时间变化的学科,即药物在体内的吸收、分布、代谢和排泄.因毒性与药物代谢动力学密切相关,药物代谢动力学是药物研发过程重要的研究内容.
药物进入体内的途径有口服、注射、吸入和渗透等.通过不同方式进入人体的药物,在体内也通过不同的方式排出.因药物在每个阶段(ADMET)都以不同的形式存在于体内并发挥作用,药物在到达靶点位置前有一定质量的损失.所以,在给药时需考虑药物的剂量和次数,这样既保证了药效,又避免了药物中毒.
ADMET预测的步骤:数据收集及描述—特征选择—模型构建—模型评价—应用域定义.人体ADMET性质的数据多,具有保密性、不易收集的特点,因此一般选择小鼠和大鼠等的数据作为参考,再与全面、准确的数据库(ToxCast,Tox21和EPA等)比较,通过数学方法对化合物结构进行特征变量描述,最后构建模型并进行评估.
肿瘤的发生和发展是个错综复杂的过程.在多种药物联合使用时,由于不同药物表现出不同的药物代谢动力学特征,临床专家需确定联合用药的时间,从而应对肿瘤治疗药物出现耐药性的情况.Chakrabarti等开发了癌症进展中药物代谢动力学与药物相互作用效应的计算框架[64],并考虑细胞克隆的预期细胞数、耐药概率和药物之间的浓度依赖性等因素.将该方法应用于TATTON临床试验,使用奥希替尼(AZD9291)和司美替尼(selumetinib)治疗表皮生长因子受体(epidermal growth factor receptor,EGFR)突变的肺癌患者,结果显示,轻微调整给药频率可显著增加药物对肿瘤细胞的抑制作用,且低于最大耐受剂量(maximum tolerable dose,MTD)的药物浓度不影响药效发挥,大大降低了药物的毒性.Demetriades等量化细胞毒性及优化肿瘤细胞增殖和凋亡影响的计算模拟方法[65],通过改变药物浓度模拟结直肠癌HCT-116细胞系和乳腺癌MDA-MD-231细胞系的多个耐药环境,并通过基于贝叶斯的模型参数优化方法,阐明多个化疗药物的治疗机制,结果表明,可通过计算机模型优化药理变量等条件定向模拟模式治疗条件.
5 结语
随着人类基因组计划的完成,蛋白组学的迅速发展,与人类疾病相关基因的大量发现,药物作用的分子靶标被相继发掘,为肿瘤的靶向治疗提供更多、更新的手段.在天然产物的开发与应用中,计算机辅助药物设计技术具有降低成本、缩短实验周期和提供导向的作用.相信随着计算机技术的发展,更先进的计算平台会提供更精准、快速及多元化的药物设计手段.
