基于PVDF-co-HFP制备具有阻酸性能的交联型阴离子交换膜的研究
2023-05-26薛上峰徐格婷祁志福刘春红高强生沈江南
薛上峰, 徐格婷, 祁志福*, 董 隽, 刘春红*, 高强生, 沈江南
(1. 浙江省白马湖实验室有限公司, 杭州 310000; 2. 浙江工业大学 化工学院, 杭州 310014;3. 工业新水源技术浙江省工程研究中心, 杭州 311121)
现代世界,对发展和增长的渴望永远不会结束.这种需求导致了工业化的快速扩张,如造纸、电镀、采矿、湿法冶金、钢铁加工等.但令人遗憾的是,在这些工业生产中排放了大量有毒的酸性废水,对环境造成了不可避免的威胁,产生了潜在的健康危害[1-3].考虑到可持续发展战略,这一废酸液可以作为一种宝贵的可回收资源.目前,人们已经广泛探索了各种分离技术来回收酸,如溶剂萃取、冷凝和蒸馏等,但由于投资成本高、能耗高,这些技术都有其局限性.为了克服这一问题,电渗析、超滤和扩散渗析等基于先进的膜分离技术已被广泛用于酸回收过程[4-14].在所有这些工艺中,扩散渗析(DD)是最可靠的处理工业废水的工艺,因为它的投资成本低,几乎为零能耗,对废水的净化效率更高,并且具有环境友好性[13-14].但是由于DD过程需要大量的水来作为酸的接收液,这会导致水资源的过量使用[11].在一些水资源短缺的环境中,很难发挥其不耗能的独特优势.并且,DD回收酸的浓度普遍较低,仍然需要后续的浓缩过程.因此,电渗析具有良好的浓缩性能是可靠的酸浓缩应用技术[15-18].但是传统的阴离子交换膜(AEM)在酸浓缩过程中存在着严重的氢离子泄露过程[19-20].相关研究表明,由于质子水化离子半径小,在电场力的驱动下,质子在溶液中的迁移速率远高于其它阳离子[21-22].“Grotthuss机制”[21]和“Vehicle机制”[22]表明,水与质子会形成团簇,质子通过分子扩散以水合分子的形式通过AEM.在后一种情况下,离子过膜时,水合离子会脱去水合层断裂形成质子和水分子,使得质子通过转运机制通过离子交换膜,导致质子的泄露.因此,迫切需要提升AEM的阻酸性能.交联作为有效的调控膜结构的手段,能够提高膜的致密度[19],有效阻碍氢离子泄露进程.
所制备的膜除了具有良好的阻酸性能外,还需要具有良好的酸稳定性.聚偏氟乙烯(PVDF)是一种良好的酸稳定性材料[23].C-F键的高离解能提供了高机械强度、良好的化学和热稳定性、优异的耐老化性和高成膜性,这些都是膜分离材料的重要特性[24-25].由于PVDF是疏水性的,需要对PVDF进行亲水性修饰才能制备出合适的AEM.这可以在制备过程中通过与亲水性改性剂[26]混合、通过脱氟化氢与适当单体接枝的化学改性[27]、轰击电子束[28]、原子转移自由基聚合(ATRP)等[29]方法实现.其中,通过脱除氟化氢形成双键后再双键共聚改性的方法操作简便且能够形成强有力的交联结构,更有利于电渗析酸浓缩的过程.从酸稳定性以及阻酸性能这两个角度来看,我们主要关注PVDF-co-HFP作为骨架,并倾向于用双键共聚接枝的手段来制备合适的AEM.
在这项研究中,开发了一种新的策略,将PVDF-co-HFP与1-乙烯基咪唑接枝.1-乙烯基咪唑有一个相互作用的胺基,可以很容易地通过季铵化或与所需分子交联来官能化.通过加入不同刚性的交联季铵化试剂形成稳定的交联结构,从而使所制备的膜具有良好的酸稳定性及阻酸性能.所制备的膜能够有效地解决传统阴离子交换膜在电渗析酸浓缩过程中氢离子泄露严重的问题,对阻酸型阴离子交换膜的制备具有一定的借鉴作用.
1 实验部分
1.1 材料
PVDF-co-HFP,美国西格玛有限公司;1,4-二溴丁烷、1,4-二溴甲基苯、4,4-二溴甲基联苯、偶氮二异丁腈(AIBN)、1-乙烯基咪唑、N-甲基吡咯烷酮(NMP)、N,N-二甲基乙酰胺(DMAC)、异丙醇、氢氧化钠(NaOH),均购自于上海阿拉丁有限公司.
1.2 PVDF-co-HFP的脱HF
PVDF-co-HFP的脱除HF过程和之前所报道的过程相类似[23].首先,称取20 g的PVDF-co-HFP颗粒,加入到300 mL的DMAC中,在磁力搅拌器上搅拌6 h,以保证充分溶解,得到无色透明溶液A;然后,配置0.15 mol/L的氢氧化钠异丙醇溶液40 mL,缓慢滴加到溶液A中并充分搅拌6 h得到深红色溶液B.紧接着,将溶液B缓慢倒入去离子水中充分洗涤,得到的淡黄色丝状固体.最后,将所得到的固体在80 ℃下真空干燥24 h.记作DPVDF-co-HFP.
1.3 阻酸阴离子交换膜的制备
首先,称取3 g DPVDF-co-HFP加入到100 mL的三口烧瓶中,并加入60 mL NMP充分搅拌3 h.紧接着,加入0.945 mL (9 mmol) 1-乙烯基咪唑和质量分数2%(0.06 g)的引发剂偶氮二异丁腈(AIBN).在加热反应之前,需要在N2氛围下通气30 min以确保排出装置里面的空气.然后,在70 ℃下油浴加热反应10 h.在未交联的铸膜液中加入3种不同的交联剂,分别加入1,4-二溴丁烷,1,4-二溴甲基苯,4,4-二溴甲基联苯各2.25 mmol.在常温下,磁力搅拌10 h至反应完全.得到的铸膜液用砂芯漏斗过滤以去除未溶解的聚合物或粉尘颗粒,过滤后得到交联的铸膜液.最后,将铸膜液倒入清洁无尘,尺寸为8 cm × 8 cm的玻璃模具上,60 ℃下真空干燥24 h,生成均质、透明、黄褐色的薄膜,分别记为AEM-1、AEM-2、AEM-3.所制备的过程如图1所示,不同膜的配料比详情见表1.
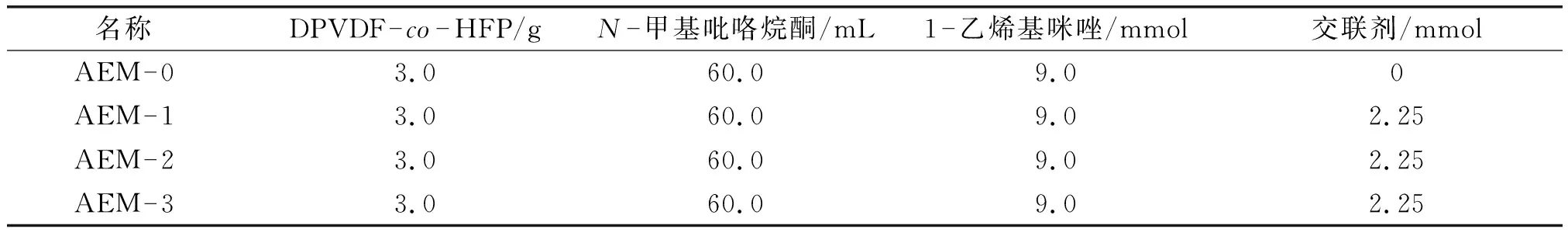
表1 所制备膜的物料配比Table 1 The composition of the as-prepared membranes

图1 AEM-x(x=1、2、3)的制备过程Fig.1 Preparation of the AEM-x(x=1,2,3)
1.4 阻酸阴离子交换膜的表征
1.4.1傅里叶红外光谱图
用傅里叶变换衰减全反射红外光谱仪(ATR-FTIP,Nicolet 6700,美国)对所制备的膜的化学结构进行红外光谱表征,来确认产物的成功合成.红外光谱的分辨率为4 cm-1,红外光谱范围设置为400~4 000 cm-1.将膜样品裁剪为1 cm×1 cm大小.送样前,膜在80 ℃下真空干燥24 h.
1.4.2扫描电子显微镜
采用场发射扫描电子显微镜(SEM,SU8010,Hitachi,日本)表征所制备交联型阴离子交换膜的微观形貌.测试前,膜在80 ℃下真空干燥24 h后,进行喷金处理.电压为10 kV.
1.4.3含水率和溶胀率
含水量(WU)通常定义为每克干膜的含水量,是评估膜导电性以及物化稳定性的重要依据.将膜置于去离子水中48 h,完全溶胀后,用滤纸擦拭膜样品以除去表面游离水,并立即用电子天平称量,获得湿膜质量(Wwet,g).相应的干膜质量(Wdry,g)可以在80 ℃下,真空干燥24 h后,用电子天平测量得到.每个样品平行测量3次,并记录平均值.
溶胀度(SR)指膜吸附溶剂分子(水分子)达到平衡时,溶胀后的体积与未溶胀前的体积的比值,衡量膜体积变化以及在实际应用中的稳定性,体现其应用前景.将膜剪成2 cm × 2 cm的方块,置于去离子水中48 h,完全溶胀后,用滤纸擦拭膜样品以除去表面游离水,再次测量其长度(Lwet,cm).
WU和SR值计算公式如式(1)、式(2)所示.
(1)
(2)
式中:Wwet为湿膜质量,g;Wdry为干膜质量,g;Ldry为干膜长度,cm;Lwet为湿膜长度,cm.
1.4.4离子交换容量和膜面电阻
使用离子交换法测定制备的AEM的离子交换容量(IEC).首先,将膜样品在60 ℃下,真空干燥24 h后称质量,记作Wdry.然后,将膜浸没在0.10 mol/L的NaCl溶液中24 h,将离子基团转换为Cl-型.紧接着,用去离子水洗涤样品表面以去除样品表面过量的NaCl溶液.最后,将膜样品浸没在0.10 mol/L的Na2SO4溶液中24 h.K2CrO4作为指示剂,用0.10 mol/L AgNO3滴定来测量释放到溶液中的氯离子的量.IEC计算公式如下:
(3)
式中:IEC为离子交换容量,mmol/g;VAgNO3为AgNO3标准溶液消耗的体积,mL;CAgNO3为AgNO3标准溶液浓度,mol/L;Wdry为干燥后离子交换膜的质量,g.
所制备膜的面电阻由图2所示装置测得.电极室用0.5 mol/L的H2SO4作为电极液在两室之间进行循环.料液室采用0.5 mol/L的H2SO4溶液作为料液,不进行循环.测试之前,将待测膜浸入0.5 mol/L的H2SO4溶液浸泡12 h,使复合膜内部的通道以及IEC内部达到离子平衡.将待测膜夹在料液室之间,将两片CMX阻酸型阳离子交换膜夹在料液室和电极室之间,两支Hg/HgSO4电极放置在待测膜表面两侧,由万用表读取电压值.空白电压的测量方法与上述步骤一致,只需去掉待测膜读取电压值即可.膜面电阻(R)计算公式如下[30-31]:
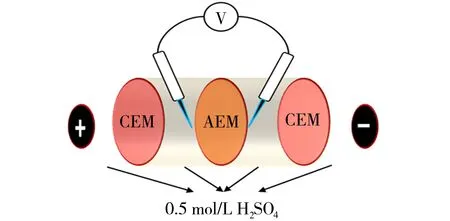
图2 膜面电阻测试装置Fig.2 Surface area resistance test device of membranes
(4)
式中:R为膜面电阻,Ω·cm2;U为膜面电压,V;U0为空白电压,V;I为恒定电流,0.05 A;S为有效测试面积,7.065 cm2.
1.4.5膜的极限电流密度
通过测量并记录待测离子膜的电流-电压曲线计算得到的极限电流密度,测试装置与离子膜面电阻的测试装置相同.测试时极液设置为0.5 mol/L H2SO4溶液,待测离子膜两侧隔室中的溶液更换为0.05 mol/L H2SO4溶液.在测试时,电流区间设置为0~0.2 A,电流梯度设置为0.01 A.每次改变电流后,通过万能表测量并记录待测膜两侧的电压.
1.4.6酸稳定性和机械强度
在60 ℃下充分浸泡在0.5 mol/L H2SO4中,测量其72 h内离子交换容量以及失重率的变化趋势.
利用拉伸电子万能试验机(CTM2050)测试膜的机械性能.将湿膜样品裁剪为1 cm×4 cm,擦干其表面残余水分,在常温下,以2 mm/min的速度测试其拉伸强度,听到“砰”声后测试终止.记录膜的断裂伸长率(Eb)和拉伸强度(TS).
(5)
式中:Eb为断裂伸长率,%;L为最终拉伸长度,cm;L0为初始长度,cm.
1.4.7酸浓缩性能测试
采用自制的四隔室电渗析浓缩酸装置进行待测阴离子交换膜的电渗析浓缩硫酸的性能,以评估制备的阴离子交换膜的电渗析硫酸浓缩性能.图3为电渗析浓缩硫酸的机理图.其中,电渗析浓缩酸装置由两个装载了钌涂层钛电极的极室、浓缩室和淡化室组成.在浓缩H2SO4测试中,电流密度设置为10 mA/cm2.极室、浓缩室和淡化室中进料的H2SO4溶液初始浓度均为0.5 mol/L.为了减少整个电渗析浓缩酸过程中因浓度差引起的水迁移导致的隔室溶液体积的变化,淡化室中硫酸溶液的初始体积为250 mL,浓缩室中的硫酸溶液初始体积为15 mL.实验过程中蠕动泵的转速设置为60 r/min,使淡化室中的H2SO4溶液保持循环.在电渗析浓缩酸过程中,在第0、10、20、30 min,1、2、3、4、5、6 h用移液枪分别对浓缩室和淡化室的H2SO4溶液取样500 μL.通过酸碱中和滴定法测试取样的溶液中H+的浓度,同时记录每次取样时刻的装置两端电压.测试结束后,准确测量并记录浓缩室和淡化室里的H2SO4溶液体积.电渗析浓缩H2SO4的能耗和电流效率由式(6)和式(7)计算得出[31]:
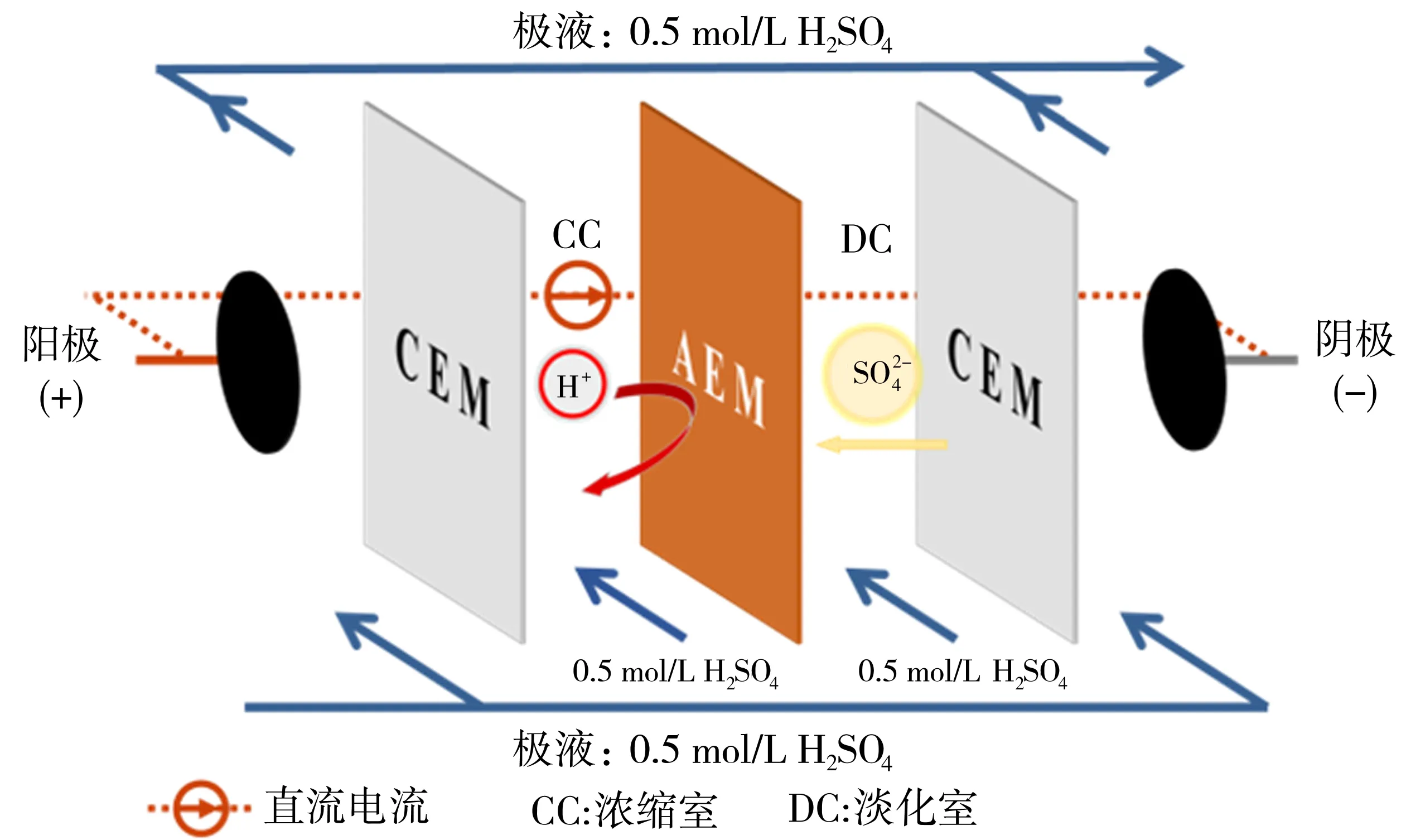
图3 电渗析浓缩硫酸机理图Fig.3 Mechanism diagram of ED of acid concentration
(6)
(7)
式中:η为电流效率,%;C0为电渗析浓缩酸过程中0时浓缩室里的H+浓度,mol/L;Ct为电渗析浓缩酸过程中t时浓缩室里的H+浓度,mol/L;Z为H+离子的价态;V0为0时浓缩室中的溶液体积,mL;Vt为t时浓缩室中的溶液体积,mL;F为法拉第常数,96 485 C/mol;I为电流密度,mA/cm2;S为离子膜的有效膜面积,7.065 cm2;E为能耗,kW·h/kg;U为电渗析装置两端的工作电压,V;Mb为H2SO4的分子量,g/mol.
2 结果与讨论
2.1 膜的化学组成

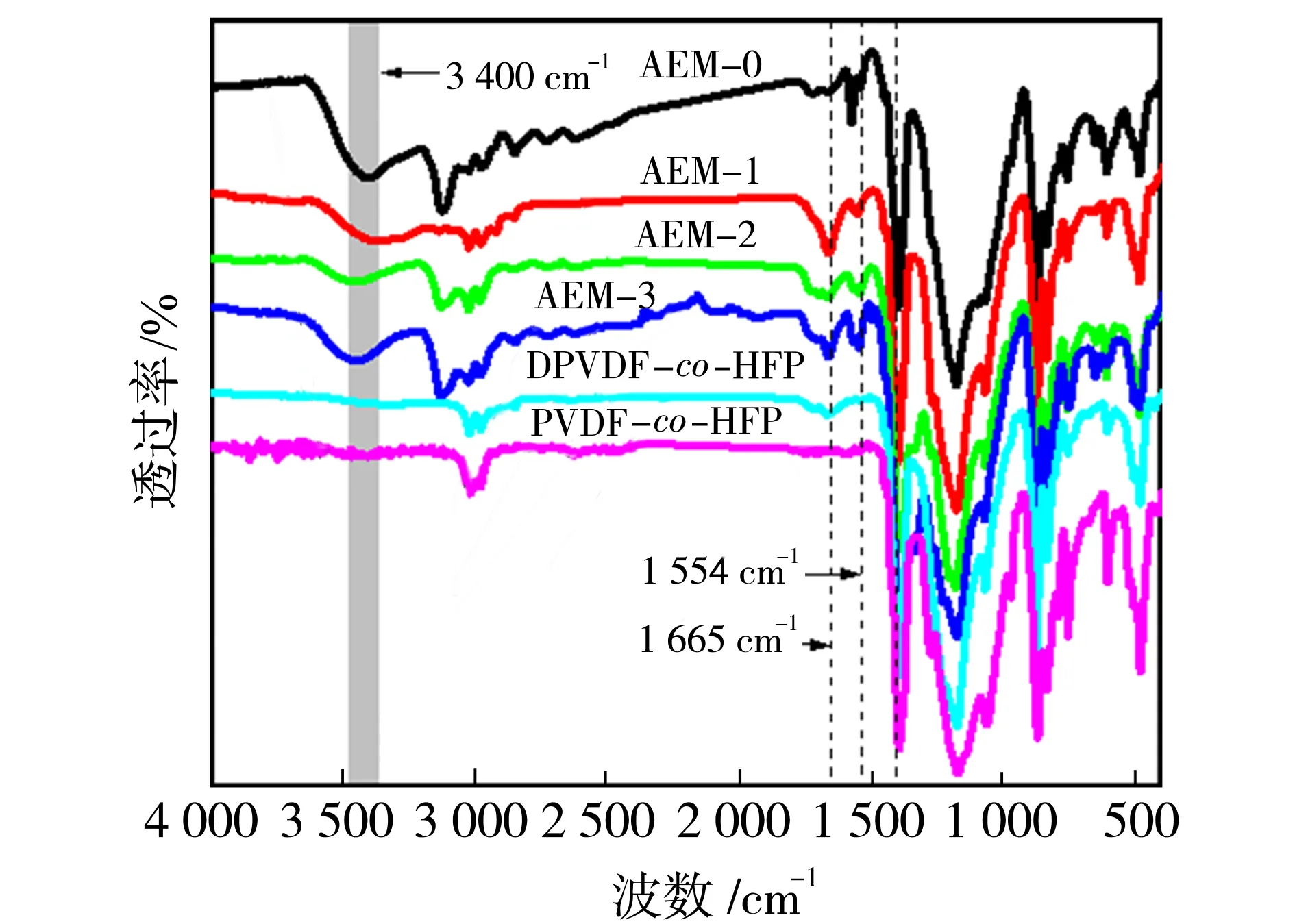
图4 PVDF-co-HFP,DPVDF-co-HFP与AEMs的红外光谱图Fig.4 FTIR-ATR spectra of PVDF-co-HFP, DPVDF-co-HFP and AEMs
2.2 膜的表面形貌
为研究离子交换膜的微观表面形貌,用扫描电子显微镜获取3种不同交联剂制成的阴离子交换膜的SEM图像,如图5所示.可见所制备的AEMs表面均无孔光滑致密.这有助于电渗析工艺浓缩酸的过程中氢离子的泄露[35].这3种膜的电镜图表面并无明显的结构上的区别.这可能是因为所加入的交联剂既是季铵化试剂的同时也是交联剂.这种交联结构会使得膜表面更加致密均匀,因此SEM图中并无太大区别.

图5 AEM-x(x=1、2、3)的SEM图像Fig.5 SEM images of the AEM-x(x=1,2,3)
2.3 膜的机械强度以及酸稳定性
离子膜的机械性能也是衡量电渗析酸回收应用中膜能否长期稳定运行的一项关键因素.图6为室温下湿态AEM-x与AHA断裂时的拉伸强度和断裂伸长率.从图可看出,AEM-1、AEM-2、AEM-3以及膜AHA的拉伸强度依次递增,断裂伸长率依次递减.这可能归因于:(1)交联剂的不同,离子膜的致密程度不同,结构的刚柔性不同.AEM-1用1,4-二溴丁烷作交联剂,离子膜结构以脂族链为主,更具柔性.而AEM-2,AEM-3分别用1,4-二溴甲基苯和4,4-二溴甲基联苯作为交联剂,分别具有1个和2个苯环,而芳香环刚性强,随着苯环增多刚性增强,因此AEM-2,AEM-3断裂伸长率降低而拉伸强度变大;(2)AHA的拉伸强度较大是由于其以织网作为支撑层.从而使得商业膜AHA具有更高的断裂强度.结果表明,加入3种交联剂制成的离子膜相比AHA具有较极好的断裂拉伸强度的同时也保持相对稳定的断裂强度.在保证优秀的力学稳定性之外,能够具有更好的加工性能,满足电渗析膜应用的要求.

图6 室温下AEM-x与AHA的断裂伸长率和拉伸强度Fig.6 Elongation at break and tensile strength of the AEM-x (x=1,2,3) and AHA under wet condition at room temperature
以PVDF-co-HFP为基膜材料的阴离子交换膜本身具有优异的耐酸性能,但是在高浓度、长时间条件下进行酸浓缩时,酸性环境可能会导致阴离子交换膜的降解与结构破坏,影响其性能.因此,耐酸性优劣关乎阴离子交换膜寿命长短,也是评价阻酸阴离子交换膜实用性能的重要依据之一,对所研发的阻酸阴离子交换膜能否投入实际应用中起到至关重要作用.如图7(a)所示,在0.5 mol/L H2SO4环境下,经过72 h耐酸性测试,AEM-3膜失重最少,但是会随着在酸性环境中时间增长失重增加;AEM-1失重明显高于AEM-2与AEM-3,但是其随时间的增长失重率没有明显变化;AEM-2的失重率与AEM-3相近,并且随时间变化平稳.但所有膜的失重率始终保持在1%左右,这可能是因为强交联结构以及C-F链的高稳定性从而使得膜的抗氧化性增强[23].这表明所制备的膜具有很好的酸稳定性.图7(b)为3种交联膜离子交换容量的变化趋势.3种交联膜的IEC变化范围分别为:AEM-1,1.23~1.20 mmol/g;AEM-2,1.28~1.24 mmol/g; AEM-3,1.24~1.20 mmol/g.随着时间变化AEM-2、AEM-3的IEC下降趋势更明显,而AEM-1的IEC变化平稳.但3张交联膜的IEC变化均在0.03~0.04 mmol/g之内,说明从IEC指数出发,交联膜的耐酸稳定性优良,其中AEM-1的稳定性更佳.综合权衡膜失重率与IEC变化趋势,在酸性环境下,交联膜具有很好的酸稳定性能,具有投入电渗析应用的潜质.
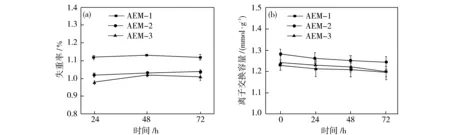
图7 AEM-x(x=1、2、3)在酸性环境下的失重率(a)与离子交换容量(b)Fig.7 The weight loss (a) and the IEC values (b) of as-prepared four AEM-x (x=1, 2, 3) in acidic condition
2.4 膜的物理性质与化学性质
离子膜吸附的水分子对于离子在离子膜中的传输具有重要作用.但吸水过多会导致离子膜过度溶胀,从而机械性能降低.实验测试了不同交联剂制成AEM与商业离子膜AHA在不同温度下的吸水率和溶胀率.不同的交联剂会显著影响吸水率和溶胀率.如图8(a)所示,比较不同交联剂膜与商业膜AHA的吸水率,从小到大依次为AHA、AEM-3、AEM-2、AEM-1.此外,随着温度从20 ℃升高到40、60和80 ℃,不同膜的吸水率也会增加,且在温度较低时(20~40 ℃)吸水率增长趋势更显著.如图8所示,离子膜的溶胀率表现出与吸水率相似的变化.与AHA相比,交联AEM-x的吸水率与溶胀率较高可能是因为季铵基团与Br-是一种亲水性较高的基团,经过咪唑化以及交联季铵化后引入了亲水性的部分,从而导致吸水率与溶胀率增加.而AHA表现出较低的吸水率和溶胀率可能是因为其具有织网作为支撑层,机械强度高,吸水性能差,且亲水性弱,因此不易溶胀.而在相同取代度的情况下,AEM-1的SR和WU依次高于AEM-2与AEM-3可能是因为三者所用交联剂的不同.从AEM-1到AEM-3,交联剂苯环数量增加,膜的刚性会随苯环的增加而增加,因此AEM-3所含苯环数量最多则其刚性最强,由此得出其WU、SR在交联AEM中最小.结果表明,具有较强的刚性结构的离子交换膜能够有效地防止膜的溶胀,从而保持稳定的膜内结构.有利于电渗析的长期稳定运行.

图8 不同温度下AEM-x与AHA的吸水率(a)与溶胀率(b)Fig.8 WU (a) and SR (b) of AEM-x and AHA at different temperatures
离子交换容量(IEC)是衡量阴离子交换膜性能优劣的一项重要指标.随着IEC的增高,则离子交换能力增强,亲水性与电导率随之升高[36].但是通常难以平衡IEC与膜机械强度之间的关系,IEC增高时其吸水率和溶胀率也随之增强,对电渗析过程产生影响[37-39].所以需要控制合理IEC值,以使膜能够同时具有较高电导率和较强膜稳定性.
由图9(a)为3种不同交联剂的离子交换容量.实验测得所制备的不同交联膜的离子交换容量与商业膜AHA相近但是略低于AHA.IEC由高到低分别为:AHA(1.29 mmol/g),AEM-2(1.28 mmol/g),AEM-3(1.24 mmol/g)和AEM-1(1.23 mmol/g).IEC的差异可能是交联结构造成的.交联会使膜结构致密紧凑,缩小离子通道从而降低IEC.又由于交联达到一定程度,膜出现微相分离结构使亲疏水相分离增强离子电导率而减弱了IEC降低趋势.微相分离与致密结构之间的平衡使IEC略低于商业膜.

图9 AEM-x和AHA的离子交换容量(a)与膜面电阻(b)Fig.9 Ion exchange capacity (a) and surface area resistance (b) of AEM-x and AHA
图9(b)为3种交联AEM与AHA膜的膜面电阻,膜面电阻从高到低分别为:AEM-1(3.5 Ω·cm2),AHA(3.4 Ω·cm2),AEM-3(2.8 Ω·cm2)和AEM-2(2.5 Ω·cm2).从图9中,可以更直观看到离子交换容量与膜面电阻之间的关系.IEC较大的膜其膜面电阻低,反之亦然.这可能是由以下两个原因造成的.(1) 可能是由于IEC较大意味着功能基团增多,为酸浓缩过程中硫酸根离子提供了更多的交换位点;(2) 可能是由于1,4-二溴丁烷,1,4-二溴甲基苯和4,4-二溴甲基联苯,这3种交联剂的刚柔性不同.苯环增多使得膜结构刚性增强,从而弱化溶胀程度,并且形成了亲疏水相分离的微相分离结构,因此便于离子透过,导致其由较低的膜面电阻.而AHA具有较高电阻的原因可能是因为AHA具有织网支撑层,该支撑层的存在使膜的结构更为致密,离子在膜内的迁移更为困难.结果表明,所制备的膜IEC以及面电阻均在电渗析要求合理的范围内.
极限电流密度(LCD)是膜能够经济运行的最大电流密度,离子膜在施加电流和电压范围内的适用性对电渗析应用至关重要[40].在0.05 mol/L H2SO4溶液中对3种交联阴离子交换膜及商业膜AHA的极限电流密度进行测试,如图10所示.电流电压曲线中欧姆区、平台区和超极限电流区的不同特征区域分别表现出反离子输运、浓差极化和过限水分裂现象.

图10 AEM-x和AHA的电流密度-电压曲线(a)和极限电流密度(b)Fig.10 Current-voltage curve (a) and limiting current density (b) of AEM-x and AHA
在欧姆区,由于外加电压增加导致离子浓度增加,因此电流密度随着外加电压增大而线性增长.并且可以根据不同AEM在欧姆区对应的斜率不同,比较其膜面电阻不同.从图10(a)中分析欧姆区斜率可初步判断商业膜AHA的膜面电阻最小,AEM-2、AEM-3的膜面电阻与商业膜AHA相近,略大于商业膜,其中AEM-1的膜面电阻与商业膜存在显著差异.随后,当电流密度接近极限电流密度时,由于淡室中的反离子浓度降低,离子供应不足.从而在平台区,改变电流密度时,电压陡增.这在电流电压图中表现为电流密度的平稳段.欧姆区与平台区切线的交点即为LCD,如图10(b)所示.可以看出商业膜AHA的LCD最大,之后依次为AEM-2、AEM-3、AEM-1,LCD分别为18.52、15.56、13.44、11.73 mA/cm2.在过限区域,电流密度再次随电压的增大而增大.因为在此电压范围,水分子发生了分裂,生成了H+和OH-.在这一阶段还会发生离子膜的浓差极化、引力对流等[41],在这一过程中电渗析的能耗将大大增加.从图10(b)可以看出,交联阴离子膜的电流密度值在11.73~15.56 mA/cm2这个范围内.因此,根据最低电流密度原则,电渗析酸浓缩过程中的电流密度应不超过11.73 mA/cm2.在电渗析的过程中,溶液中离子迁移的速度低于电解水产生OH-与H+的速度会导致浓差极化,因此为保证较高的电流效率并防止浓差极化以及溶质电解产生较高能耗,选择在极限电流密度以下进行测试.本实验选择10 mA/cm2的极限电流密度.
2.5 膜的酸浓缩性能
使用AHA、AEM-1、AEM-2、AEM-3进行电渗析酸浓缩测试.H2SO4溶液的初始浓度设置为0.05 mol/L,淡室中硫酸溶液的初始体积为250 mL,浓缩室中硫酸溶液的初始体积为15 mL,电流密度设置为10 mA/cm2.实验结果如下所示.
图11展示了4种膜在电渗析酸浓缩过程中浓缩室中H+的浓度随时间的变化趋势.图中可以明显看出,浓缩室中的H+浓度随运行时间的增加而显著增加,而淡化室中的H+浓度基本保持不变.在电渗析酸浓缩运行3 h后,商业膜AHA浓缩结果接近极限,增长趋势减慢,而交联膜则在4 h后才出现减缓的增长趋势,交联改性膜浓缩极限均明显大于AHA膜.浓缩极限由大到小依次为:AEM-3>AEM-2>AEM-1>AHA.在这3种交联型阴离子膜中,AEM-3将H+从0.1 mol/L浓缩至0.74 mol/L,酸浓缩能力最优;其次为AEM-2,浓缩至0.68 mol/L;最后为AEM-1,浓缩至0.64 mol/L.三者酸浓缩能力的差异可能是由于以下几个原因造成.(1) 离子交换容量不同.AEM的阻酸性能会随着膜IEC的增大而降低.(2) 是通过交联形成了微相分离结构,微相分离结构不仅有助于削弱质子泄露,还有助于削弱水的迁移[42].(3) 是由于吸水率和溶胀率的不同.许多文献证实WU和SR会对质子的运输产生影响,其中低WU和SR有利于防止质子阻塞[43-44].造成差异的根本原因可能是使用交联剂的不同,致使所制成的膜的结构、性能等存在差异.由图9可知,AEM-2与AEM-3的离子交换容量均大于AEM-1.又根据图8推断,可能是由于在咪唑功能化以及交联后季铵化之后,提高了膜的含水率和溶胀率,一定程度限制了AEM-1的质子传输.因此,低IEC,高WU、SR的AEM-1酸浓缩能力低于AEM-2、AEM-3.AEM-3酸浓缩极限与AEM-2相比较高可能是因为AEM-3为联苯结构,由图6可知其刚性强则结构更稳定,对膜结构具有更好的稳定支撑作用,不易在酸环境下因为溶胀而使得膜结构发生变化.可知AEM-3的溶胀率与含水率在3种交联膜中最低,也有利于阻止H+的反向扩散运输.综上所述,AEM-3的酸浓缩效果最优.
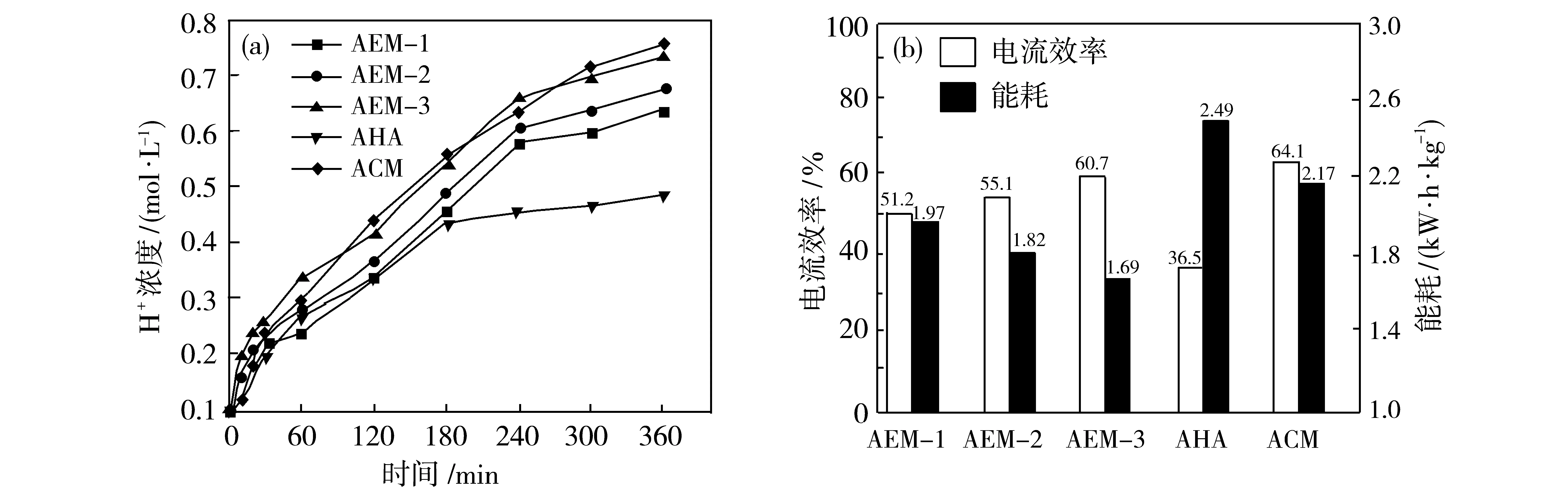
图11 AEM-x和AHA以及ACM的酸浓缩性能(a)以及电流效率和能耗(b)Fig.11 The current efficiency and energy consumption of AEM-x and AHA, as well as ACM
根据酸浓缩的极限计算其电流效率.由式(6)和式(7)可知,控制一定电流密度、有效膜面积以及酸浓缩时间,电渗析酸浓缩的电流效率随浓缩室中H+的浓度增高而增高.实验结果测得电流效率从大到小依次为:AEM-3(η=60.7%)>AEM-2(η=55.1%)>AEM-1(η=51.2%)>AHA(η=36.0%).AEM的膜面电阻是影响电渗析过程能耗的重要因素之一[44].由图9(b)可知,AEM-1的膜面电阻最高而AEM-2与AEM-3的膜面电阻均明显低于AEM-1及AHA.据报道,电流效率可作为评估阴离子膜阻氢性能的特征指标[40].当膜面电阻较高时,则离子不易透过离子交换膜,需消耗更多能量以通过相同数量的离子.因此,可认为AEM-2与AEM-3具有优异的阻氢能力,且AEM-3与其他离子膜相比,具有最高的电流效率与最低的能耗,分别为60.7%和1.69 kW·h/kg,且拥有最高的酸浓缩极限.同理,可能是由于AHA膜不具有阻酸性能,因此虽然其膜面电阻低于AEM-1,但是其电流效率低于交联膜导致最终酸浓度低从而引发能耗高.
对商业膜ACM的酸浓缩数据进行了单独的阐述.由于商业阻酸膜ACM上的功能基团为弱碱基团,因此其在电渗析运行过程中能耗相对较高.达到2.17 kW·h/kg,相较于AEM-3能耗增加了28.40%.与此同时,弱碱基团的存在使得ACM膜具有更低的含水率从而能够提升膜的阻酸性能.最终酸浓度达到0.76 mol/L,相较于AEM-3,0.74 mol/L,略微较高.从而电流效率也相对较高[从式(6)可以得知电流效率跟最终酸浓度成正相关].综上所述,从能耗和酸浓度方面考虑,发展能耗更低的交联型离子交换膜在酸浓缩过程中具有更好的应用前景.
3 结论
为指导解决现今工业废酸处理问题,本工作制备一种以PVDF-co-HFP为基膜材料的交联耐酸阴离子交换膜,并利用电渗析技术以达到回收废酸使其资源化利用这一目的.所制备的AEM-x均在保持合适的离子交换容量的同时具备优良的机械强度,优化平衡了阴离子交换膜的含水率和溶胀率,具备可观的耐酸性能.1,4-二溴丁烷交联膜、1,4-二溴甲基苯和4,4-二溴甲基联苯分别能将0.1 mol/L的H+浓缩到0.64、0.68、0.74 mol/L,证明经过交联改性的AEM具有良好的酸浓缩性能.其中AEM-3具有突出的综合性能,与传统商业膜对比性能优越.为解决电渗析回收酸过程中质子泄露这一问题具有一定的借鉴意义.
