夏季珠江口冲淡水对氨氧化古菌的空间演替及分布规律的影响
2023-05-23荆红梅周鹏张玥刘皓刘红斌
荆红梅,周鹏,张玥,刘皓,刘红斌
(1.中国科学院 深海科学与工程研究所,海南 三亚 572000;2.南方海洋科学与工程广东省实验室(珠海),广东 珠海 519082;3.中国科学院三亚海洋科学综合实验室,海南 三亚 572000;4.香港科技大学 海洋科学系,香港 999077)

研究者主要采用16S rRNA基因分析方法进行不同环境样品中AOA的丰度以及系统发育分析[4].但该基因过于保守,甚至不同群落的16S rRNA基因相似性能达97%以上,所以很难将AOA的不同种属在16S rRNA基因水平上区分开来[5].随着分子生物学技术的发展以及对AOA的深入研究,发现了氨氧化过程中的关键酶—氨单加氧酶(ammonia monooxygenase,AMO).该酶是由amoA、amoB、amoC基因分别编码的α、β、γ 3个亚基组成的三聚体膜结合蛋白,属于单加氧蛋白家族[6].相比较16S rRNA基因作为分子标记,编码α亚基的amoA基因具有较强的保守性和明显的专一性,在相近的群落之间具有高分辨率,并且具有功能基因特性,因而在氨氧化古菌的遗传多样性分析方面具有优势,常被作为研究AOA的分子标记[7].
目前利用AOA功能基因amoA和qPCR技术来研究不同生境中AOA的基因丰度分布特征的报道较多,包括中国内蒙古草地、东北的海河、珠江口到南海的沉积物、日本海、北太平洋加利福尼亚海湾和蒙特利海湾等.不同水体环境中的AOAamoA基因丰度也不同.通常海水样品中的AOAamoA基因拷贝数介于103~108copies/L之间;且表层海水的丰度普遍低于透光层区域;不同营养类型湖泊中基因拷贝数为7.80×104~1.34×106copies/L[8],珠江底泥中的拷贝数为9.60×109~5.10×1010copies/kg[3].
珠江年径流量约3 492亿m3,仅次于长江,是黄河年径流量的6倍.全长2 320 km,流域面积约44万km2,且4至9月的径流量占全年的80%.珠江口是三角洲网河和残留河口湾并存的河口,径流大,潮差小,含沙量相对较小.河口区河汊发育,水网密布[3].此外,珠江口夏季受冲淡水影响明显,流速大.目前对于珠江口AOA的研究表明其在水体中的丰度通常高于氨氧化细菌[9],以Shallow group簇和SCM1-like簇样亚系为优势类群,且在DNA水平上的整体类群组成与cDNA水平上活跃参与氨氧化过程的类群具有明显差异[10].同时AOA在较大的颗粒有机物上表现出更高的转录活性[11].为了深入了解夏季珠江口冲淡水对功能微生物类群的影响,利用新一代高通量测序技术和荧光定量PCR(qPCR)技术研究了夏季珠江口不同盐度、不同深度及不同过滤孔径(0.22 μm 代表自由生活的;3 μm代表附着的)的各水体样品中的AOA群落结构组成以及丰度分布特征.此外,在DNA和cDNA水平上进行了比较研究,解析了关键的生态因子,系统阐述径流输入对AOA的群落演替的影响.
1 材料和方法
1.1 样品采集
珠江口夏季海水样品借助厦门大学航次于2014年7月9日至26日采集.共包括9个站位(图1):A02、A04、A06、A08、A10、A12、A14、A16、A18.每个站位的表层及底层海水经3 μm和0.22 μm的聚碳酸酯滤膜来分级过滤,以收集附着生活(3 μm,attached)的和自由生活(0.22 μm,free-living)的微生物类群.滤膜放入加有500 μL RNAlater的冻存管中并立即冻入液氮罐中,回到实验室后保存于-80 ℃超低温冰箱中.
1.2 核酸提取,PCR扩增与测序
滤膜上的DNA采用Genomic DNA Mini Kit试剂盒提取,RNA采用PureLinkTM RNA Mini Kit试剂盒提取,然后第一链cDNA采用SuperScript®Ⅲ First-Strand试剂盒合成.利用含MID的特异性引物对amoAF和amoAR扩增AOAamoA基因序列[12].用无菌水作为反应的阴性对照.DNA使用1 μL为模板,cDNA使用3 μL为模板;每个PCR的反应体系总体积为20 μL.PCR反应程序为:95 ℃预变性5 min;95 ℃变性45 s,53 ℃退火1 min,72 ℃延伸1 min,运行35个循环;最后72 ℃延伸7 min,保存在4 ℃.PCR产物用质量分数1.2%的琼脂糖凝胶进行检测,以100 bp Ladder为DNA标准Marker.PCR产物经纯化后,使用Roche GS Junior测序平台进行高通量测序.
1.3 实时荧光定量PCR
AOAamoA基因的定量PCR反应采用amo196F和amo277R[8]为引物,利用SYBR®Premix Ex TaqTM试剂盒,在ABI 7500 Fast定量PCR仪上进行.每个qPCR的反应体系为10 μL,其中1 μL DNA或cDNA为模板,并使用无菌水作为阴性对照,每个样品和质粒均做3个平行.实时定量PCR反应程序为:95 ℃预变性10 min;95 ℃变性15 s,60 ℃延伸1 min,45个循环;然后进行溶解曲线检测阶段,荧光取值在60 ℃和溶解曲线全程.利用梯度稀释的质粒(10-8~10-1)分别作为模板进行qPCR,并用其分别对应的Ct值和基因拷贝数来构建标准曲线(R2=0.990 6,扩增效率为102.16%),然后将样品(DNA、cDNA)通过qPCR获得的Ct值分别代入标准曲线中进行计算,最后用SigmaPlot对数据进行绘图.
1.4 生物信息学分析
通过Roche GS Junior平台所获得的AOAamoA基因原始序列利用mothur软件(http://www.mothur.org/)进行初步筛选和去杂,将引物以及测序标签的错配碱基数大于1的序列去除,同时将总序列长度小于300 bp的序列去除,随后去除含有嵌合体的序列,最终形成一个shared文件.利用mothur的get.oturep命令基于97%的序列相似度得到各个OTU的代表序列名.随后,按照样品最低序列为标准对数据进行均一化处理.利用Primer 5进行多样性指数分析,主要包括群落丰富度指数(Margalef)、均匀度指数、香农指数、辛普森指数等,其计算公式分别为:d[Species Richness(Margalef)]=(S-1)/ln(N),J'(Pielou's Evenness)=H'/ln(S)、H'(Shannon)=-∑i[Pi×ln(Pi)],1-λ'(Simpson)=1-∑i[Ni×(Ni-1)/N×(N-1)],其中S为群落总OTU数,N为总序列数,Pi为第i个OTU在整个序列中所占的比例,Ni为第i个OTU的个体数.各站位序列覆盖度(Coverage,C)是利用mothur软件的summary.single命令基于97%的序列相似度对氨氧化古菌amoA基因序列进行计算的,其计算公式为C=[1-(n1/N)],其中n1为序列中仅有一条序列的OTU数,N为序列的总OTU数.
利用mothur软件进行样品间相似度的聚类分析,其中非度量多维尺度分析(NMDs)利用Primer 5[13]完成.此外,还利用mothur的tree.shared命令基于布雷柯蒂斯(Bray-Curtis)距离进行样品间的非加权组平均法(UPGMA)聚类分析.
挑选丰度较高的OTUs(总序列数大于20)进行系统发育分析.先将这些OTUs的代表序列上传到NCBI的GenBank数据库(http://blast.ncbi.nlm.nih.gov/Blast.cgi)做blastn比对,并下载参考序列.基于ClastalW进行序列比较,并切除两端不匹配的序列,然后利用Mega 6[14]的Models进行模型预测.挑选出最优的模型General Time Reversible,利用最大似然法(Maximum Likelihood)进行Bootstrap值为1 000的系统发育树构建.
1.5 统计学分析
利用CANOCO V5.0对氨氧化古菌的群落结构组成和环境因子之间进行冗余分析(RDA);并利用SPSS 20对amoA基因丰度和主要类群与环境因子之间分别进行两两之间的Spearman相关性分析,以确定达到极显著水平(p<0.01)或显著水平(p<0.05)的环境生态因子.
1.6 基因序列的提交号
将获得的原始序列提交至NCBI数据库,获得序列号PRJNA948716.
2 结果与讨论
2.1 氨氧化古菌群落多样性
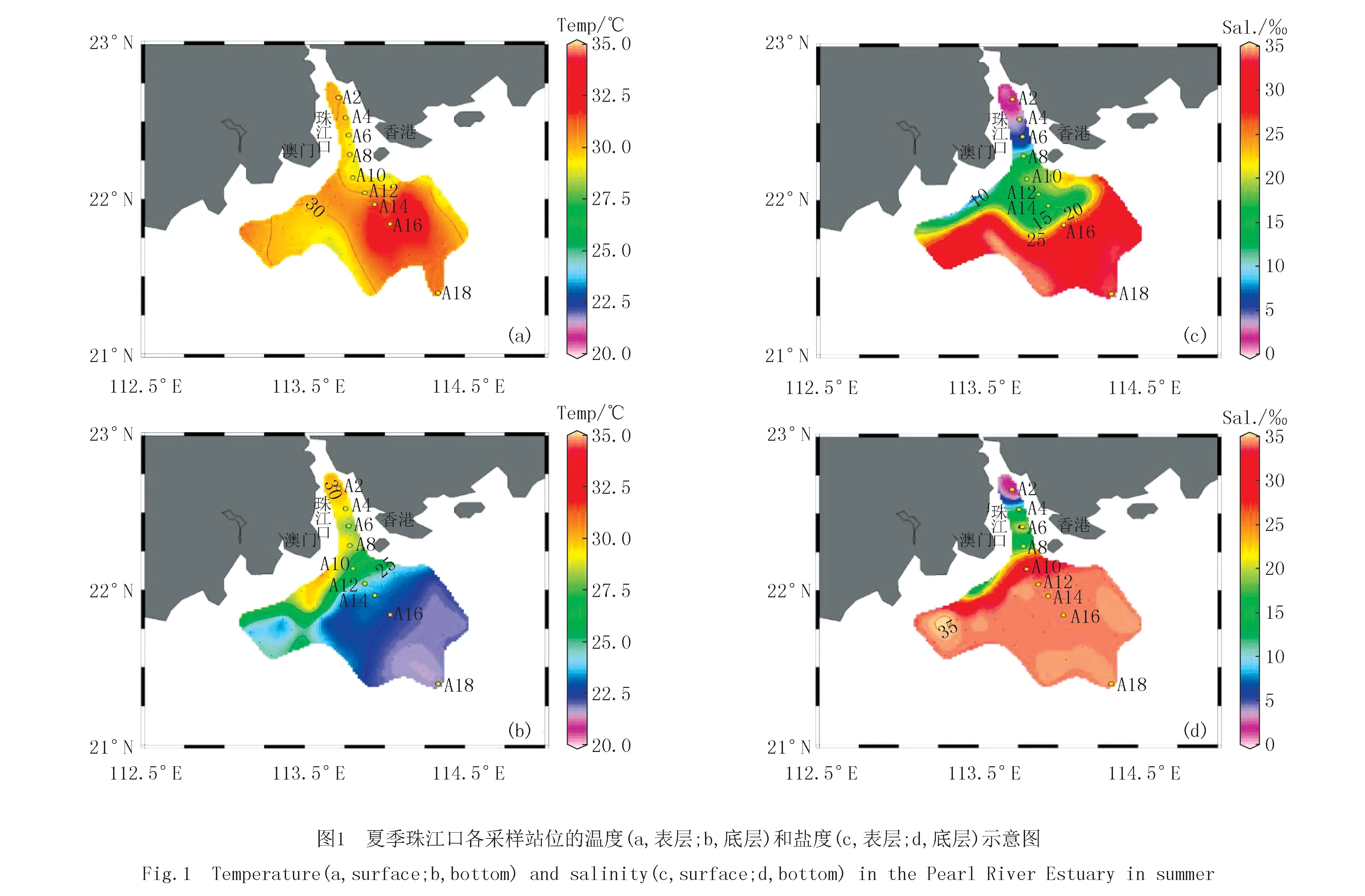
夏季珠江口的表层及底层的温盐差异非常明显(图1).从珠江河口A2一直到外海A18的断面的盐度差异尤其明显,包括了全淡水环境到淡海水交汇环境最后到全海水环境(图1).
每个站位的样品序列的覆盖度均达到了0.99(表1),且稀释曲线随测序量增加而逐步达到饱和,均显示了测序深度足够反映样品的多样性.

表1 夏季珠江口氨氧化古菌amoA基因测序信息
总体而言,夏季珠江口各站位AOA的OTUs偏低,数目最多的A2B-0.22拥有52个OTUs,而A18B-3仅含有6个OTUs(表1).其次,底层样品的OTUs数目高于表层;cDNA样品的OTUs数目高于DNA,结果与之前的研究相似[10].主要是淡水来源的A2B站位含有最大的OTU数(52),同时也有最大的群落丰富度指数(6.34)、均匀度指数(0.56)、香农指数(2.22)和辛普森指数(0.81)(表1).生物群落的多样性反映了生物群落的丰富度以及各个类群的相对比例.群落丰富度指数(Margalef)越高,说明此环境中生物群落的种类越丰富;对于均匀度指数J'(Pielou's Evenness)来说,指数越高说明在此环境中各群落的分布均一性越好;辛普森指数代表了群落优势度,其值越高说明群落在此环境种类越丰富;香农指数则是综合考虑了群落丰富度和均匀度的一个群落多样性指数,其指数值越高,则反映在此环境中群落多样性越高.基于各个参数,大概得出以下结论:(1)盐度较低的站位(例如A2、A4、A6和A8),AOA的群落多样性显著高于其他盐度较高的站位(p<0.05);(2)同一研究站位,表层来源的AOA群落多样性高于底层;(3)通过DNA水平和cDNA水平的比较,发现cDNA水平上的群落多样性反而高于DNA水平;(4)cDNA水平的均匀度指数高于DNA水平,说明AOA的一些丰度较低的物种却具有极强的转录活性或者一些丰度较高的物种具有极低的转录活性,如丰度最高的OTU1在DNA水平上的相对比例为68%~97%,cDNA水平上为48%~60%;而OTU2在DNA水平上的相对比例为2%~27%,cDNA水平上为25~45%.cDNA水平上更为直观地反映了活跃代谢的微生物类群,能增加人们对于微生物群落组成和分布及生态功能的全面认识,如之前在珠江口的研究发现某些SCM1-like簇亚系的高转录活性[10].此外,转录水平上的结果还需要结合其他研究手段来反映真正的生态功能.
2.2 AOA amoA基因的系统发育分析
夏季珠江口的AOA主要聚于7簇,分别是Shallow group簇,Freshwater簇,SCM1-like簇,Coastal簇,Sediment簇,Deep group簇,Soil簇,其中Shallow group簇被细分为Shallow group Ⅰ亚簇、Shallow group Ⅱ亚簇,Shallow group Ⅲ亚簇;Freshwater簇和Coastal簇是本研究中新发现的并分别命名(图2).

Shallow group簇是夏季珠江口的优势种群,这与之前的研究相一致[10].其中Shallow group Ⅰ亚簇包括OTU1和OTU3在内共7个OTUs;Shallow group Ⅱ亚簇也包含了8个OTUs;Freshwater簇,SCM1-like簇,Coastal簇,Sediment簇,Deep group簇,Soil簇分别包含8,3,6,4,2,2个OTUs.这些序列绝大多数来源于海洋水体,少部分序列来自海洋沉积物和陆地生态系统.其中Shallow group Ⅰ亚簇的同源序列来源于中国东海水体和南海水体、沉积物[15];Shallow group Ⅱ亚簇的同源序列来源于扬子江河口[16]、东北太平洋的缺氧区[12]、东海水体和南海水体及沉积物[15],同时CN25菌株[17]也聚集在这个亚簇;Freshwater簇的同源序列来源于东江水体[18]及淡水水族馆[19];Coastal簇的同源序列包括2株海岸分离的纯菌(PS0和HCA1)[2]和来自墨西哥海湾、南海表面沉积物[16]的环境样品序列;Sediment簇的同源序列来源于夏威夷珊瑚礁保护区沉积物、加利福尼亚湾沉积物[12];Deep group簇的同源序列来源于东北太平洋缺氧区、加利福利亚湾沉积物[9]和东海水体[15];Soil簇的同源序列来源于崇明岛东滩湿地[20]、太湖沉积物[21]和东海水体[15].
2.3 氨氧化古菌群落结构组成
通过对得到的总计185个OTUs进行分类鉴定,获得珠江口夏季AOA的群落结构组成信息(图3).

除A2B-0.22样品外,其他样品都被Shallow group Ⅰ亚簇主导,而A2B-0.22样品的AOA来源主要是Freshwater簇,其中底层样品来源的OTU1和表层样品来源的OTU3分别在其对应的群落结构组成中占据着较大比例,而OTU1和OTU3在系统进化树上均属于Shallow group Ⅰ亚簇.在Tara Oceans的研究中,Shallow group Ⅰ亚簇在全球海洋的表层AOA群落中占主要地位[22],这表明Shallow group Ⅰ亚簇更适合此水域的环境,正如其代表性培养种(CandidatusNitrosopelagicusbrevisCN25)的基因组和蛋白质组学所揭示的结果,Shallow group Ⅰ亚簇具有高编码密度和普遍分布于寡营养表层海洋的流线型基因组[23].通过对同一站位样品DNA和cDNA水平上进行比较,Shallow group Ⅱ亚簇在cDNA水平上拥有相对更高的比例;Deep group簇只在cA16B-0.22和cA18B-0.22两个样品中存在;且Soil簇来源的也主要在A2B-0.22样品中发现.本研究的群落结构组成与南海海域100 m[15]的AOA群落结构组成极为相似,但本研究中未发现Shallow group Ⅲ亚簇.这可能是由于本研究的采样点集中在珠江口,而Shallow group Ⅲ亚簇主要在南海开放海域中发现所造成的.
2.4 氨氧化古菌群落聚类分析
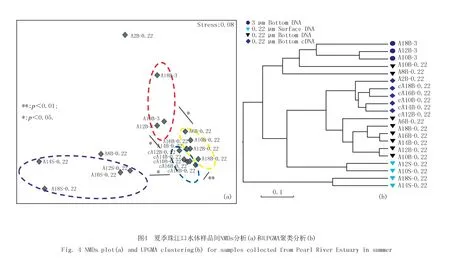
通过NMDs和UPGMA两种聚类分析,样品间的相似性聚类表现出了几乎一致的结果(图4):除淡水来源A2B站位和盐度相对较低的A8B站位外,所有底层海水来源的0.22 μm(free-living)样品聚集在一起;表层海水来源的free-living样品聚集在一起;而底层海水来源的3 μm(attached)样品聚集在一起,说明表层和底层来源的AOA差异较明显.自由生活的与附着的AOA群落之间也存在着一定的差异,但相对于表层和底层之间的差异较小.
2.5 氨氧化古菌的amoA丰度分布
利用标准曲线分别计算自由生长和附着生长的AOAamoA基因拷贝数(图5).自由生活(0.22 μm)的AOAamoA在DNA水平上表层拷贝数为2.31×102~2.76×105copies/L,其中A2、A8、A16站位未检出;底层AOAamoA基因拷贝数为3.40×103~1.81×106copies/L;而底层cDNA水平上AOAamoA基因拷贝数为4.66×101~1.73×104copies/L且A2和A6站位未检出.附着生活(3 μm)的AOAamoA在DNA水平上表层拷贝数为8.57×101~1.98×104copies/L,其中A8站位未检出;表层cDNA水平上AOAamoA基因拷贝数为1.93×102~5.87×103copies/L且A2、A8、A16和A18站位未检出;底层AOAamoA基因拷贝数为2.21×102~6.60×103copies/L;而底层cDNA水平上AOAamoA基因拷贝数为1.06×102~5.81×103copies/L.通过以往的文献报道显示:珠江表层水体中AOAamoA拷贝数为 6.27×104~3.63×107copies/L,而底层水体为3.59×105~4.98×108copies/L[10];不同营养类型湖泊中AOAamoA基因拷贝数为7.80×104~1.34×106copies/L[8].总体而言,0.22 μm比3 μm有着更高的amoA基因丰度;无论是在0.22 μm孔径还是在3 μm孔径上,amoA的丰度都有着从河口到外海升高的趋势;而从DNA相对于cDNA上的占比看,0.22 μm孔径上的比值大于3 μm孔径,说明自由生活的AOAamoA基因活性比附着生活的低,表明了AOA微生物的生活策略与之前的研究相符[10].在0.22 μm孔径上,底层的样品的amoA基因丰度远远大于表层.

2.6 相关性分析
RDA分析表明夏季珠江口氨氧化古菌群落组成受到温度和盐度显著影响.Spearman相关性分析显示,温度与盐度及Shallow group Ⅱ亚簇呈极显著负相关(p<0.01);盐度与Shallow group Ⅱ亚簇呈极显著正相关(p<0.01),而与SCM1-like簇呈负相关(p<0.05);但温度和盐度均未与amoA基因丰度显示出任何相关性(表2).以往对珠江口河口到南海沉积物中氨氧化微生物的研究也证明了盐度是影响AOA群落结构的关键因素,且AOA的丰度与pH和温度呈正相关[3];珠江口沉积物中的AOA丰度与盐度相关,且盐度越低丰度更高[24].而本研究发现AOAamoA基因丰度随着盐度降低而降低,这可能是本研究聚焦于海水样品所造成的差异,且河口系统中AOA的amoA基因丰度不仅仅受盐度因子的调节,同时还会受到温度、无机营养盐浓度和溶解氧的组合控制[11].
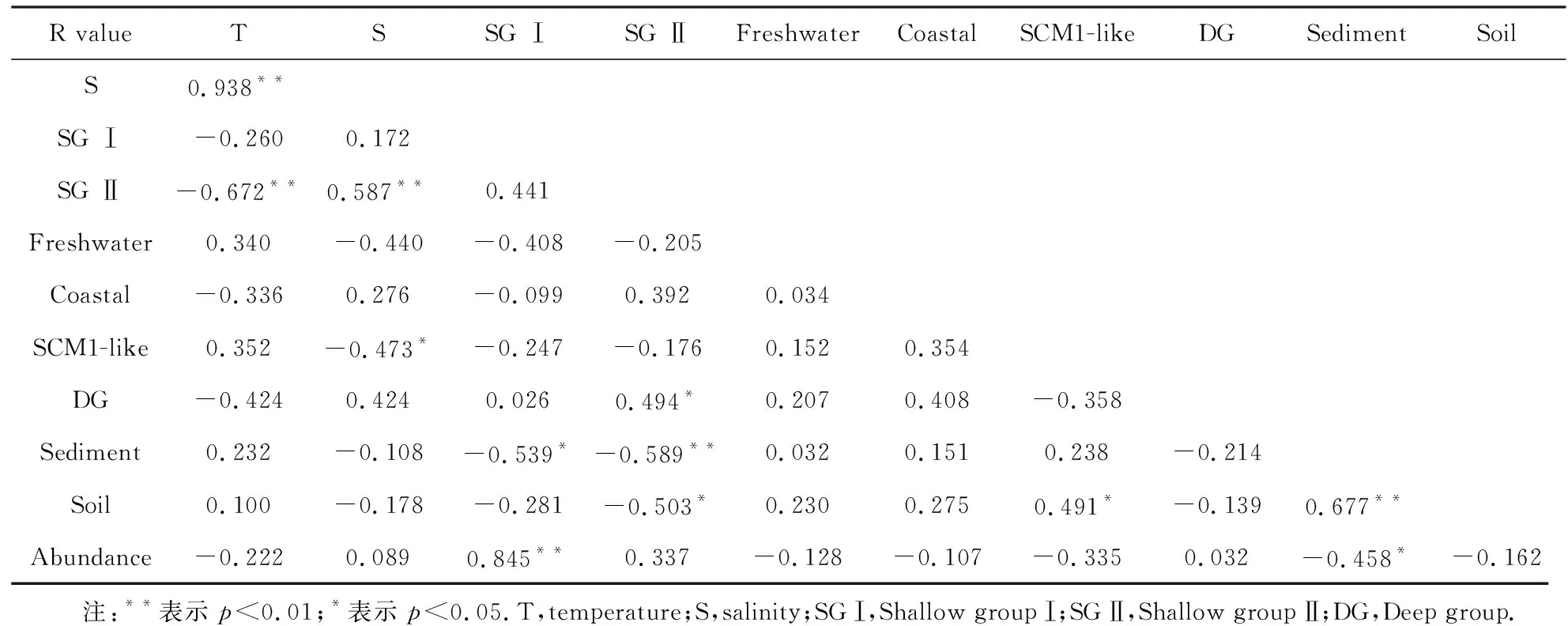
表2 夏季珠江口氨氧化古菌群落组成、丰度与环境因子之间的Spearman相关性分析
3 结 论
通过对夏季珠江口氨氧化古菌的群落结构及丰度分布进行研究,研究结论如下:(1)主要是淡水来源的A2站位有着最大的AOA群落多样性但有最低的amoA丰度,且群落结构组成主要是Freshwater簇占主导;(2)除A2站位外,其他样品均被Shallow group Ⅰ亚簇主导,而Shallow group Ⅱ亚簇有着更高的转录活性;(3)AOAamoA基因丰度在底层样品中明显高于表层,且free-living的AOA丰度比attached高10~1 000倍;(4)Shallow group Ⅱ亚簇与温度存在极显著负相关,但其和SCM1-like簇与盐度存在着显著正相关.
