基于PacBio 平台的黄颡鱼全长转录组测序及分析
2023-05-09王家琪熊阳韩庆庆皇培培梅洁
王家琪,熊阳,韩庆庆,皇培培,梅洁
(华中农业大学水产学院,武汉 430070)
近年来,转录组学技术广泛应用于水产动物繁育、营养、发育和免疫等各研究[1]。目前,转录组测序应用最广的是二代测序技术(RNA-Seq),二代转录组测序具有测序通量高、成本低的优势。尽管二代测序技术读取准确率高,但读长相对较短,给后续序列组装、拼接以及注释等带来困难[2]。而基于PacBio 平台的单分子实时测序技术(Single molecule real time,SMRT)的第三代全长转录组测序技术,在测序过程不需要打断RNA 片段,可超长读取包含单个完整转录本序列信息、后续无需序列拼接与组装,极大地提高基于功能注释的准确性[3]。此外,三代全长转录组测序可直接读取目标序列,无须PCR 扩增等步骤,大大降低了假阳性率,同时也避免偏置及碱基替换等问题,精准度高达99.9%[3,4]。
黄颡鱼(Pelteobagrus fulvidraco)是中国一种重要的小型淡水经济鱼类,肉质鲜美且营养价值高。经全国水产原种和良种审定委员会审定的黄颡鱼新品种有全雄黄颡鱼“全雄1 号”(GS-04-001-2010)和杂交黄颡鱼“黄优1 号”(GS-02-001-2018)[5,6]。遗传育种技术的创新和新品种的培育推动了黄颡鱼产业的发展,2019 年黄颡鱼年产量高达53.69 万t[7]。随着研究技术的不断革新,关于黄颡鱼分子遗传育种的研究逐渐增多。从传统的基因克隆、单核苷酸多态性(SNP)[8]、简单重复序列(SSR)[9]和扩增片段长度多态性(AFLP)[10]等分析,到转录组[11]、基因组[12]和蛋白质组[13]等组学大数据分析,为黄颡鱼种质资源挖掘和遗传育种奠定了基础。关于黄颡鱼不同组织的二代转录组分析在近些年也有不少报道,冯美惠等[14]对饲料中添加维生素D3的黄颡鱼肠和肾脏组织进行转录组分析;Wu等[15]对XY 黄颡鱼和YY 超雄黄颡鱼的精巢组织进行转录比较分析;Chen等[11]对黄颡鱼的卵巢、精巢、肝脏、肾脏、肌肉、脑、脾和心脏8 种组织进行454 焦磷酸测序法混样测序分析。目前在黄颡鱼中还没有第三代全长转录组测序结果的报道。
本研究采用PacBio 平台的第三代测序技术对黄颡鱼10 种组织的RNA 混样进行全长转录组测序,测序结果与黄颡鱼基因组进行对比分析,挖掘出新基因和已知基因的同源异构体,进行序列分析、功能注释和基因结构分析,为黄颡鱼分子遗传育种提供科学理论。
1 材料与方法
1.1 组织样本采集
三龄性成熟的黄颡鱼购买于武汉百瑞生物有限公司。使用MS-222 将黄颡鱼麻醉后,分离出肝脏、肾、背部肌肉、脑、脾、心脏、皮肤、血液、鳃、性腺等组织,用液氮速冻后送至武汉菲沙基因信息有限公司进行RNA 提取、质量和浓度测定及测序分析。
1.2 文库构建
利用Trizol 法提取黄颡鱼组织的总RNA。通过Nanodrop 检测RNA 的纯度(OD260nm/280nm)和浓度,Agilent 2100 对RNA 的完整性进行精确检测;使用琼脂糖凝胶电泳检测有无基因组DNA 污染。以上各组织RNA 检测合格后进行等量混匀,使用SMARTer PCR cDNA Synthesis Kit 合成全长cDNA,全长cDNA片段通过BluePippin 筛选共获得3 个文库(1~2 kb,2~6 kb(a)和2~6 kb(b));通过PCR技术对全长cDNA进行扩增;对全长cDNA 进行末端修复,加上SMRT哑铃型接头和使用核酸外切酶消化;通过BluePippin 进行二次筛选,获得测序文库。使用Qubit 2.0 和Agilent 2100 对构建的文库进行质量检测,检测结果达到要求后进行上机测序。
1.3 全长转录组测序数据分析
使用PacBio 平台对检测合格的文库进行测序。对测序下机原始输出数据使用SMRT Link v5.0 进行处理,获得Subreads,对单分子多测序序列进行自我纠错处理,获得环形一致性序列(Circular consensus sequence,CCS)。通过检测确定CCS 序列包含5′端引物、3′端引物以及poly-A 后进行分类,找出全长非嵌合(Full-length non-chimeric read,FLNC)序列。采用GMAP 软件[16],将FLNC 序列对比至黄颡鱼基因组上[12],再根据每条FLNC 序列的比对位置,统计分析基因座(loci)和转录本异构体(isoform)。另外,通过冗余转录本的去除和低可信度转录本的过滤获得合格的isofrms。将测序得到的loci 和isoform 与参考基因组注释的loci 和isoform 进行比较,可以确定检测到已知基因新的isoform 以及鉴定到新基因的isoform。测序得到的基因满足以下任一条即判定为新基因:①与已注释基因没有overlap 或overlap 小于20%;②与已注释基因overlap 大于20%,但基因方向不一致。将本次测序获得的转录本和参考基因组注释得到的转录本进行比较分析,如果参考基因组注释的基因转录本与三代转录组测序分析的isoform不同时为单外显子,或转录组测序分析的isoform 存在1 个以上新的剪切位点,则认为该同源异构体是新的同源异构体。
1.4 IncRNA 预测和基因结构分析
将新基因的isoform、已知基因的新isoform 序列与NR、KOG、KO 库比对,过滤掉潜在的编码序列;对于在NR、KO 和KOG 库中没有hit 的序列,进一步利用CNCI、CPC2、CPAT 和PLEK 评估序列的编码潜能,过滤编码潜能大于设定的cutoff 或长度<200 bp的序列,取4 个软件预测结果的交集序列,作为最终的非编码RNA 预测的结果[17,18]。PacBio 长读长测序实现了全长转录本测序。相对于二代短读长RNA-Seq 测序识别可变剪接时完全依赖于junction reads 比对的方法,三代全长测序使得直接基于全长isoform 序列相互比较的可变剪接识别成为可能。用ASprofile 软件对测序得到的isoform 可变剪接事件分别进行分类和统计[19]。融合基因通过以下方式被确定:定位到2个或2个以上的远距离范围,定位比对至少占转录物的10%,覆盖率≥99%,每个定位位点必须至少相距100 kb[20]。利用全长转录组APA 检测软件Tapis进行可变多聚腺苷酸化位点检测[21]。
1.5 新基因功能注释
使用以下公共数据库对新基因进行基因功能注释:非冗余蛋白数据库(Non-redundant protein database,NR)、蛋白质真核同源数据库(Eukaryotic orthologous groups,KOG)、基因本体论数据库(Gene ontology,GO)、东京基因与基金组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)和蛋白质序列数据库(Swiss-Prot)。
2 结果与分析
2.1 测序结果与数据分析
全长转录组测序获得9 525 639 个Subreads 和842 830 个环形一致性序列(CCS)Reads,其中,带有5′端引物的reads 数有755 439个,有3′端引物reads数有772 185个,有Poly-A 的reads 数有760 944 个;全长(Full-length)reads 数共693 262个,全长非嵌合(Full-length non-chimeric read,FLNC)reads数有685 574个,全长非嵌合reads 平均长度为2 736 bp,全长非嵌合N50 为3 067 bp(表1)。采用GMAP 软件将全长转录组FLNC 序列与黄颡鱼基因组对比分析,基因组已注释的loci 和isoform 均为24 552个,而基于PacBio 测得的FLNC 序列筛选后与基因组对比分析,鉴定出26 664 个loci 和72 509 个isoforms,isoforms 平均长度为2 918 bp。PacBio 分析的全部isoform 和基因组已注释isoform 的长度分布和同一loci内isoform 的个数如图1a、图1b 所示。将72 509 个isoforms 与参考基因组比较分析发现,13.01%的isoforms(9 437 个)为已知基因isoforms,69.76%的isoforms(50 580 个)为已知基因新isoforms,17.23%的isoforms(12 492个)为新基因isoforms(图1c)。

图1 isoforms 统计和分类

表1 reads分类统计
2.2 LncRNA 分析
利用CNCI、CPC2、PLEK 和CPAT分别预测了6 497、6 482、6 390、3 379 个LncRNA,并将4 个软件预测的交集部分(3 169 个LncRNA)作为最终的非编码RNA 预测结果(图2a)。根据LncRNA 在基因组上相对于蛋白编码基因的位置分为4 种:正义链(sense)有861 个、反义链(antisense)658 个、内含子间(intronic)746 个、基因间(intergenic)904 个(图2b),其所在的位置与其功能有一定的相关性。LncRNA 的长度分布见图2c,最小长度为213 bp,最大长度为5 458 bp,平均长度为2 220 bp,序列的正态分布表明LncRNA 的序列质量较好。

图2 黄颡鱼长链非编码RNA 分析
2.3 可变剪接事件、可变多聚腺苷酸化位点和融合基因分析
分析的可变剪接类型共有45 873个,分为以下几种类型:外显子跳跃(Exon skip,ES)有9 437个,占比20.57%;内含子保留(Retained intron,RI)有10 360个,占比22.58%;可变供体位点(Alternate donor site,AD)有2 182个,占比4.76%;可变受体位点(Alternate acceptor site,AA)有4 140个,占比9.03%;其他类型可变剪接形式有19 754个,占比43.06%(图3a)。利用Tapis 软件测到20 774 个polyA 位点来源于11 651 个基因(图3b),共分析出4 881 个存在可变多聚腺苷酸化位点。此外,共检测到304 个融合基因,其中,265 个融合基因来源于不同染色体上不同基因的融合,39 个融合基因来源于同一染色体上不同基因的融合(图3c)。

图3 全长转录本基因结构分析
2.4 新基因功能注释
已发现的12 492 个新基因与公共数据进行功能注释,成功注释了7 233 个isoforms,其中,非冗余蛋白(NR)数据库、基因本体论(GO)数据库、东京基因与基金组百科全书(KEGG)数据库、蛋白质真核同源(KOG)数据库和蛋白质序列(Swiss-Prot)数据库分别注释到7 224、4 291、3 865、2 477、3 641 个新基因,还有5 259 个新基因未注释上(图4)。利用GO数据库对4 291 条新基因进行注释,并分类到生物学过程、细胞组分和分子功能,其中,细胞过程(Cell process,2 345 个)、细胞组分(Cell,1 979 个)和结合功能(Binding,2 195 个)分别在三大类中数量最多(图5)。3 865 个新基因参与KEGG 代谢通路,并富集到细胞过程(Cellular processes)、环境信息处理(Environmental information processing)、遗传信息处理(Genetic information processing)、新陈代谢(Metabolism)和生物体系统(Organismal system)上,其中,运输和分解代谢(Transport and catabolism,343 个)、信号转导(Signal transduction,712 个)、折叠分类和降解(Folding,sorting and degradation,194 个)、脂质代谢(Lipid metabolism,138 个)和免疫系统(Immune system,372 个)分别在五大类中数量最多(图6)。
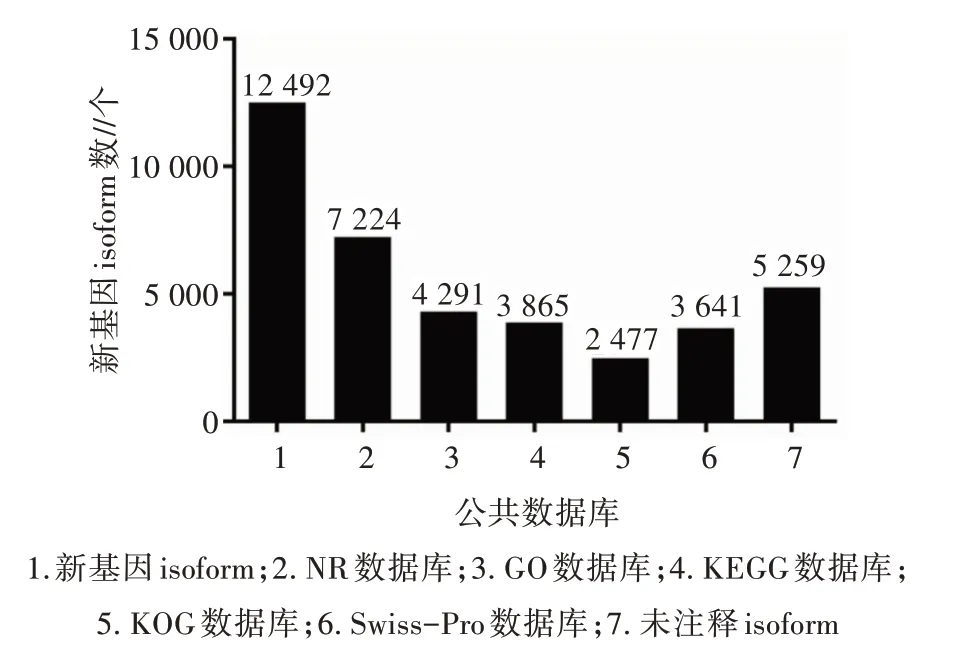
图4 全长转录本新基因注释

图5 GO 注释分类
此外,涉及黄颡鱼生殖与繁殖相关的内分泌系统代谢途径包括催产素信号通路(Oxytocin signaling pathway,70 个)、雌二醇信号通路(Estrogen signaling pathway,50 个)、孕酮介导的卵母细胞成熟(Progesterone-mediated oocyte maturation,44 个)、促性腺激素释放激素信号通路(GnRH signaling pathway,30 个)和卵巢类固醇合成(Ovarian steroidogenesis,6 个)(图7)。

图7 KEGG 注释的内分泌系统相关基因
3 小结与讨论
在过去的研究中,由于缺乏黄颡鱼基因组信息,关于黄颡鱼遗传和生理相关研究受到限制,只能通过操作繁琐、效率较低的cDNA 末端快速克隆技术(RACE PCR)获得黄颡鱼部分基因转录本序列[22,23]。随着高通量测序技术的快速发展,二代转录组测序技术广泛应用于黄颡鱼的研究[11,14,15]。由于缺乏基因组信息和二代转录组测序长度短等限制,绝大多数已发表的黄颡鱼转录组均通过无参分析,导致基因注释困难,对基因的可变剪切、融合基因和基因家族不能准确地检测[1,24]。
本研究基于PacBio 平台的单分子实时测序技术对黄颡鱼的肝脏、肾、背部肌肉、脑、脾、心脏、皮肤、血液、鳃和性腺等组织进行混样测序,共获得全长非嵌合reads 数685 574个,全长非嵌合reads 平均长度为2 736 bp;相比二代测序技术获得的黄颡鱼转录本长度大幅提升,如Chen等[11]基于454 GSFLX 测序平台获得的黄颡鱼混样转录组unique sequences 平均长度仅601 bp,而Wu等[15]和Zhu等[25]基于Illumina 测序平台的unigenes 平均长度分别为944 bp 和716 bp。LncRNA 是长度大于200 bp 的长链非编码RNA,在生物体内广泛存在,并介导许多复杂的生命活动过程[26,27]。在本研究中,利用CNCI、CPC2、PLEK 和CPAT 软件预测到3 169 个LncRNA。在此基础上,挖掘LncRNA 与内分泌和生殖过程的相关性,对其展开功能研究,对探索黄颡鱼内分泌及生殖过程具有重要意义。基因结构如可变剪接事件(AS)、可变多聚腺苷酸化位点(APA)和基因融合分析可增加转录多样性和基因功能复杂性[28-30]。有些基因的前体mRNA(pre-mRNA)通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA 剪接异构体,该过程称为可变剪接(或选择性剪接)(Alternative splicing,AS)。Weirather等[31]证明PacBio 测序平台比二代转录组测序技术更有利于AS 事件的鉴定。在本研究中,共挖掘到45 873 个可变剪接事件。Poly-A 位点的改变也是一类重要的RNA 转录后调控修饰,产生具有不同UTRs 和编码序列的mRNAs,其功能与选择性剪接相似[32]。在本研究中,共挖掘到4 881 个基因存在可变多聚腺苷酸化位点。融合基因是指来源于不同基因的2 个片段被拼接在一起形成的新基因[33]。导致2 个基因发生融合的机制包括基因组结构变异、转座或基因转录后的反式剪接等。在本研究中,共检测到304 个融合基因,其中,265 个融合基因来源于不同染色体不同基因融合,39 个融合基因来源于同一染色体不同基因融合。
目前,新品种全雄黄颡鱼“全雄1 号”和杂交黄颡鱼“黄优1 号”已成为中国主流养殖品种。由于全雄黄颡鱼均为雄性、杂交黄颡鱼性腺退化无法繁殖、大型湖泊禁捕以及“长江十年禁渔计划”等因素,导致中国黄颡鱼母本资源短缺,苗种供应不足。人们利用鱼类性逆转技术结合黄颡鱼性别连锁分子标记,成功将XX 雌性黄颡鱼逆转为XX 雄性黄颡鱼,然后XX 雄性和雌性黄颡鱼繁殖后获得黄颡鱼全雌配套系[34-36],黄颡鱼全雌配套系还需要进一步提升其繁殖性能。Hu等[37]发现部分雌性黄颡鱼存在排卵障碍问题,在常规催产药物中加入鲤脑垂体提取物可顺利排卵。23 号染色体上miR-200 簇敲除的斑马鱼存在输卵管发育缺陷和排卵障碍问题,注射催产素(OXT 和AVT)+hCG 或鲤脑垂体提取物均可以促使排卵[38],表明催产素在鱼类的排卵过程中起着重要作用。本研究挖掘到的2 477 个新基因通过KEGG 分析富集到269 个已知途径中,其中,292 个基因与动物内分泌系统相关,70 个基因富集到催产素信号、50 个基因富集到雌二醇信号通路、44 个基因富集到孕酮介导的卵母细胞成熟、30 个基因富集到促性腺激素释放激素信号通路和6 个基因富集到卵巢类固醇合成。这些数据为以后的黄颡鱼生殖和繁殖相关机制研究提供了科学依据。
