X连锁肾上腺脑白质营养不良1家系报告并文献复习
2023-04-29姜荆靳阳吴玉娇刘学伍
姜荆 靳阳 吴玉娇 刘学伍

[摘要] 目的 分析1例X連锁肾上腺脑白质营养不良(X-linked adrenoleukodystrophy,X-ALD)患者家系中发现的ATP结合盒超家族D亚家族1(ABCD1)基因突变位点,结合患者家系临床资料及相关文献,探讨X-ALD的诊断、治疗及预后。方法 回顾性分析2021年11月就诊我院神经内科的1例X-ALD患者的临床资料及其家系情况,并检索国内外相关文献进行复习讨论。结果 患者主要表现为皮肤发黑、头晕头痛、全身不适、乏力、疲惫、低血压、食欲减退、睡眠欠佳等肾上腺皮质功能减退症状及双下肢轻瘫的神经系统异常。其父母、一兄一姐、其兄之子及其姐之子均未见相关临床表现。患者颅脑及脊髓MRI检查未见异常,基因检测示患者ABCD1基因存在一处半合子突变(c.1628C>T),其母ABCD1基因存在杂合突变,其父ABCD1基因未见异常。结论 X-ALD存在多种类型,可出现肾上腺皮质功能减退及中枢神经系统异常症状,本例X-ALD患者为肾上腺脊髓神经病型,ABCD1基因第6号外显子的1处突变(c.1628C>T:p.P543L)为本患者致病原因,MRI在X-ALD的诊断中或无决定性作用。
[关键词] 肾上腺脑白质营养不良;ATP结合盒转运体,亚家族D,成员1;突变
[中图分类号] R586.9;R394
[文献标志码] A
ANALYSIS OF THE FAMILY OF A PATIENT WITH X-LINKED ADRENOLEUKODYSTROPHY AND LITERATURE REVIEWJIANG Jing, JIN Yang, WU Yujiao, LIU Xuewu (Cheeloo College of Medicine, Shandong University, Jinan 250012, China)
[ABSTRACT] Objective To analyze the mutation site in the ATP-binding cassette subfamily D member 1 gene (ABCD1) in the family of a patient with X-linked adrenoleukodystrophy (X-ALD), and to explore the diagnosis, treatment, and prognosis of X-ALD based on the patient family clinical data and related literature. Methods The clinical data and family information of a patient with X-ALD admitted to the Neurology Department of our hospital in November 2021 were retrospectively analyzed, and relevant literature worldwide was retrieved for review and discussion. Results The main symptoms of the patient were skin blacke-ning, dizziness, headache, general discomfort, fatigue, low blood pressure, loss of appetite, and poor sleep and other symptoms associated with adrenocortical insufficiency, as well as nervous system abnormalities such as lower limb palsy. His parents, brot-her, sister, son of brother, and son of sister had no related clinical manifestations. Magnetic resonance imaging of the brain and spinal cord showed no abnormalities. Genetic testing showed that the proband had a hemizygous mutation in ABCD1 (c.1628C>T), the mother had a heterozygous mutation in ABCD1, and the father had normal ABCD1. Conclusion There are many types of X-ALD with adrenocortical hypofunction or central nervous system abnormal symptoms. In this case, the patient shows the adrenomyeloneuropathy form of X-ALD. A mutation in the exon 6 of ABCD1 (c.1628C>T:p.P543L) is the cause of the disease. Magne-tic resonance imaging may not play a decisive role in the diagnostic of X-ALD.
[KEY WORDS] Adrenoleukodystrophy; ATP binding cassette transporter, subfamily D, member 1; Mutation
肾上腺脑白质营养不良(Adrenoleukodystrophy,ALD)是一种遗传代谢性疾病,所有ALD患者均存在ATP结合盒超家族D亚家庭1(ABCD1)基因突变,使得极长链脂肪酸(VLCFAs)异常累积,特别是在脑白质、脊髓及肾上腺中,出现弥散性神经脱髓鞘和肾上腺皮质功能不足的临床表现;95%的患者为男性,而女性该基因多数为杂合子,属于本病突变基因的携带者。男性ALD发病率为1/21 000~1/15 500,而男性ALD和女性杂合子携带者的共同发病率约为1/17 000[1]。本文报告了1例X连锁ALD(X-ALD)患者,该患者存在明显的肾上腺皮质功能不足及神经系统异常表现,但其颅脑磁共振(MRI)检查未见异常,对该患者及其家系进行分析,并结合文献进行复习,探讨X-ALD的诊断、治疗以及预后。
1 临床资料
患者,男,37岁,因“皮肤发黑30年,双下肢无力2年余”入院。患者5岁左右皮肤发黑明显、易感冒。7年前症状加重,逐渐出现头晕头痛、全身不适、乏力、疲惫、低血压、食欲减退、睡眠欠佳等症状,当地医院诊断为肾上腺功能减退症,口服醋酸氢化可的松片(每日2片)治疗后症状明显好转。2年前出现双下肢无力、行走不稳,症状进行性加重,伴蹲起困难,于外院内分泌科就诊考虑肾上腺相关遗传类疾病,故行基因检测。患者未婚未育,父母及一兄一姐均身体健康。入院查体:胸前区及背部见散在多发丘疹,偶感瘙痒、触痛,全身皮肤发黑。神经系统查体:记忆力、计算力、定向力等高级智能粗测正常,伸舌偏左,余脑神经检查正常;双下肢肌张力偏高,双上肢肌力5级,双下肢肌力4级;共济运动正常,行走呈痉挛样步态,深浅感觉未见明显异常,双下肢腱反射(),双侧髌阵挛及踝阵挛均(+),双上肢腱反射(),双侧霍夫曼征、巴氏征及查多克征(+),余病理征未引出,脑膜刺激征(-)。颅脑、颈胸髓MRI检查:颅脑平扫未见异常,胸椎管蛛网膜下腔见异常信号,考虑脑脊液波动伪影可能,C2、C3终板变性。肾上腺薄层CT检查示双肾上腺萎缩。肌电图示双下肢周围神经损害,脱髓鞘伴轴索损害。实验室检查结果显示:促肾上腺皮质激素(ACTH)98.725 pmol/L,皮质醇0.006 μg/L,24 h尿17-羟类固醇19.36 mg,24 h尿17-酮类固醇10.5 mg,24 h尿皮质醇164.25 μg。脑脊液常规及生化检查:颅内压220 mmHg,氯离子132 mmol/L,球蛋白阳性,葡萄糖、蛋白定量、免疫球蛋白等指标均未见异常。因技术原因及患者拒绝活检,缺少患者VLCFAs生化指标及脑组织、肾上腺、周围神经等的病理活检结果。根据患者病情调整醋酸氢化可的松片服用剂量及频率(每次半片,每8 h 1次),因患者双下肢肌张力高,行走困难,添加巴氯芬片(每次1片,每日2次)改善肌张力障碍,同时嘱患者出院以后定期随访。
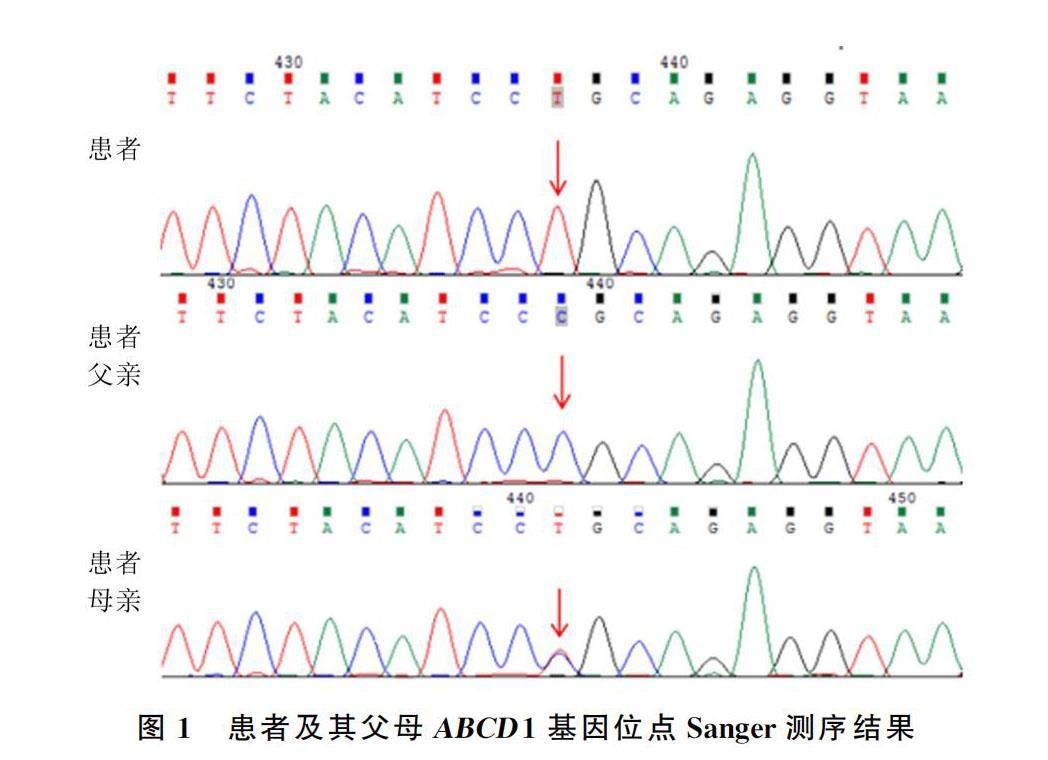
患者ABCD1基因检测第6号外显子存在一处突变(c.1628C>T:p.P543L),为半合子突变,患者父亲ABCD1基因无突变,母亲ABCD1基因为杂合突变(图1)。此外患者(2代患者5)有一姐(2代2)一兄(2代3),姐兄各育有一子(3代1、2),以上4人均无相关临床表现,患者目前未婚未育(图2)。
2 讨论
X-ALD是一种遗传代谢性疾病,致病基因是位于染色体Xq28上的ABCD1基因[2],该基因长度为21 kb,由10个外显子组成,编码745个氨基酸组成的ATP-结合盒(ABC)超家族中D亚家族的肾上腺-脑白质营养不良蛋白(ALDP)[3]。ABCD1基因突变致其表达的ALDP功能异常,该蛋白是一种过氧化物酶体跨膜蛋白,参与将VLCFA-CoA合成酶导入或锚定到过氧体膜中,并可能促进VLCFAβ氧化-过氧体和线粒体之间的相互作用。ALDP功能异常可减少β氧化,使得VLCFAs不能转膜进入细胞溶酶体进行脂肪酸氧化而异常累积,导致血浆和组织中饱和的未分支VLCFAs水平增高。特别是在脑白质、脊髓及肾上腺中,ALDP功能异常在导致细胞和血浆VLCFAs水平升高的同时,使患者出现弥散性神经脱髓鞘和肾上腺皮质功能不足的临床表现[4]。X-ALD的临床表现多种多样,并且男性患者高于女性。本例患者的基因突变类型为第6外显子c.1628C>T改变,X-ALD数据库(https://adrenoleukodystrophy.info/)中可见48例相同突变家系;截至2022年6月,数据库中共累及1 272种突变位点,但迄今尚未发现ABCD1基因突变型与该疾病表型之间的相关性。在查阅的相关文献中,存在一个错义突变P484R的家系病例报道,该家系中6例男性患者存在5种不同临床表型[5]。
根據临床表现、发病年龄及疾病进展,X-ALD目前可分为7种类型,即儿童脑型、青少年脑型、成人脑型、肾上腺脊髓神经病型(AMN)、单纯肾上腺皮质功能减退型、无症状型及女性杂合子型。其中脑型最为严重,表现为大脑脱髓鞘改变,这与脑白质中的炎症反应相关,患者主要出现认知功能减退的临床表现,多见于儿童期(儿童脑型),少见于青春期或成年期(青少年脑型及成人脑型)。儿童脑型最为多见,患儿在4~8岁起病,首发症状通常是认知功能障碍,表现为学习成绩下降和行为问题;随后可能出现局灶性神经功能障碍,如视力和听力下降,运动症状如偏瘫、构音障碍和吞咽困难,有时还会出现癫痫发作[6-7];临床表现为进行性加重,最终导致失明、耳聋或完全瘫痪。有的患者表现为大脑强直状态,最终死于中枢性呼吸衰竭、脑疝或感染等[1,8]。青少年脑型通常10~20岁起病,临床表现与儿童脑型相似但进展缓慢。而成人脑型为20岁后起病,表现为颅内病变进展迅速。单纯肾上腺皮质功能减退型通常只存在肾上腺皮质功能减退症状,不累及神经系统,但其中大部分会进展为AMN[9-10]。AMN占X-ALD表型40%~46%,发病年龄大多在3~50岁[11],进展缓慢,主要累及脊髓和周围神经,可表现为下肢进行性痉挛性截瘫、括约肌功能紊乱和性功能障碍[1]。无症状型即为患者无相应临床表现却通过检验发现VLCFAs升高,或者通过基因检测存在ABCD1基因突变。女性杂合子型是指ABCD1基因突变的女性携带者出现有AMN临床表现,大约50%的女性携带者在中年期或更晚时会出现下肢进行性痉挛性截瘫等AMN相关症状,而大脑受累和肾上腺皮质功能不全较罕见[6]。有研究报道,儿童ALD最常出现脑型,而成人ALD最常出现AMN,至少20%的AMN患者发病较晚,可发展为脑部受累,且与儿童脑型具有相同的快速进展过程[6]。本家系患者虽缺少VLCFAs生化指标检查及相关病理活检,但其以肾上腺皮质功能减退表现起病,而后累及神经系统,出现双下肢无力、行走痉挛样步态等特征性表现,依据所有基因检测结果,可以诊断为X-ALD中的AMN,其母则为ABCD1基因突变携带者,尚无症状表现。
诊断X-ALD需从ACTH实验室检查、VLCFA测定、影像学表现和基因诊断等多方面综合考虑。VLCFAs是特异性诊断标志物,但其对预测疾病发展及严重程度没有帮助[12]。而颅脑MRI是目前最主要的预测疾病进展并判断疾病严重程度的辅助检查方法,对于无神经症状的患者更应进行连续的颅脑MRI检查[13-14]。值得特别指出的是,该例患者虽然存在下肢轻瘫及肾上腺皮质功能受损变性,但颅脑及脊髓MRI均未见脑白质相关病变。根据2012年X-ALD指南[15],X-ALD典型影像学改变为双侧顶枕区白质内对称分布的蝴蝶状长T1长T2信号影,增强扫描可见病灶周围呈镶边样强化,随疾病进展病灶累及范围可由脑组织后部向前部扩展[14]。一项长期随访研究显示,27%~63%的AMN患者会出现大脑受累症状(如认知下降、行为异常、视力减退、听觉受损或癫痫发作),37%~41%的患者颅脑MRI检查发现大脑脱髓鞘改变[16]。10%~20%的成年男性大脑受累表现为MRI对比增强,且患者神经功能迅速减退,伴有严重的认知和行为障碍,可能导致患者完全残疾和过早死亡[16]。
与其他遗传代谢疾病类似,X-ALD目前没有很好的治疗手段,且多种对症治疗方法疗效欠佳[17]。对于肾上腺功能不全的X-ALD患者,美国相关指南建议ACTH大于300 μg/L需糖皮质激素替代治疗,而ACTH在100~300 μg/L之间的患者在进行兴奋试验后,若血皮质醇仍小于180 μg/L,也可应用糖皮质激素替代治疗[18-19]。该疾病的治疗方法包括早期异体造血干细胞移植[20-21]、造血干细胞基因治疗[22]、洛伦佐油疗法[15,23],近年来出现的抗氧化相关研究展现了本病治疗的新方向[24],组蛋白去乙酰化酶抑制剂(如丙戊酸钠)、氘修饰的R-吡格列酮(PXL065)[25]具有减少细胞氧化损伤、神经保护的作用,对X-ALD具有良好的治疗潜力。
本研究报道了1例ABCD1基因突变所导致的X-ALD患者,该患者虽诊断为X-ALD的AMN型,但其MRI检查结果为阴性。目前该病无有效治疗方法,早期影像学诊断有助预测预后,但不能因影像学结果阴性而排除诊断,且MRI检查在X-ALD的诊断中是否起决定性的作用还有待于进一步研究。X-ALD需要根据患者临床表现及ACTH实验室检查、VLCFA测定结果、影像学检查和基因检测结果等综合判断。X-ALD患者需调整日常饮食,降低富含VLCFA的饮食摄入,再结合他汀类药物降低体内VLCFA水平;针对肾上腺皮质功能减退应及时给予糖皮质激素治疗;早期儿童脑病可考虑异体造血干细胞移植治疗,而成人治疗方案仍有待探索。
作者声明:姜荆、靳阳、吴玉娇参与了研究设计;姜荆、靳阳、刘学伍參与了论文的写作和修改。所有作者均阅读并同意发表该论文,且均声明不存在利益冲突。
[参考文献]
[1]杨立英. 我国首部罕见病诊疗指南发布[J]. 中国卫生画报, 2019(3):64.
[2]MOSSER J, DOUAR A M, SARDE C O, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters[J]. Nature, 1993,361(6414):726-730.
[3]罗晓妹,刘丽英,邹丽萍,等. X-连锁肾上腺脑白质营养不良表型及基因型研究[J]. 中华神经科杂志, 2021,54(7):686-692. [4]MANOR J, CHUNG H, BHAGWAT P K, et al. ABCD1 and X-linked adrenoleukodystrophy: A disease with a markedly variable phenotype showing conserved neurobiology in animal models[J]. J Neurosci Res, 2021,99(12):3170-3181.
[5]BERGER J, MOLZER B, FA? I, et al. X-linked adrenoleukodystrophy (ALD):A novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes[J]. Biochem Biophys Res Commun, 1994,205(3):1638-1643.
[6]KEMP S, PUJOL A, WATERHAM H R, et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: Role in diagnosis and clinical correlations[J]. Hum Mutat, 2001,18(6):499-515.
[7]ASHRAFI M R, AMANAT M, GARSHASBI M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies[J]. Expert Rev Neurother, 2020,20(1):65-84.
[8]MA C Y, LI C, ZHOU X Y, et al. Management of adrenoleukodystrophy: From pre-clinical studies to the development of new therapies[J]. Biomed Pharmacother, 2021,143:112214.
[9]ENGELEN M, KEMP S, POLL-THE B T. X-linked adrenoleukodystrophy: Pathogenesis and treatment[J]. Curr Neurol Neurosci Rep, 2014,14(10):486.
[10]SCHFER L, ROICKE H, FISCHER M, et al. Cognitive functions in adult-onset phenotypes of X-linked adrenoleukodystrophy[J]. Ann Neurol, 2021,90(2):266-273.
[11]SMITH K D, KEMP S, BRAITERMAN L T, et al. X-linked adrenoleukodystrophy: Genes, mutations, and phenotypes[J]. Neurochem Res. 1999,24(4):521-535.
[12]RATTAY T W, RAUTENBERG M, S?HN A S, et al. Defining diagnostic cutoffs in neurological patients for serum very long chain fatty acids (VLCFA) in genetically confirmed X-Adrenoleukodystrophy[J]. Sci Rep, 2020,10:15093.
[13]LIBERATO A P, MALLACK E J, AZIZ-BOSE R, et al. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy[J]. Neurology. 2019,92(15):e1698-e1708.
[14]MALLACK E J, TURK B R, YAN H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines[J]. J Inherit Metab Dis, 2021,44(3):728-739.
[15]ENGELEN M, KEMP S, VISSER M D, et al. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management[J]. Orphanet J Rare Dis, 2012,7:51.
[16]ZHU J, EICHLER F, BIFFI A, et al. The changing face of adrenoleukodystrophy[J]. Endocr Rev, 2020,41(4):577-593.
[17]BERGER J, PUJOL A, AUBOURG P, et al. Current and future pharmacological treatment strategies in X-linked adrenoleukodystrophy[J]. Brain Pathol, 2010,20(4):845-856.
[18]ZHU J, EICHLER F, BIFFI A, et al. The Changing Face of Adrenoleukodystrophy[J]. Endocr Rev. 2020,41(4):577-593.
[19]YU J Y, CHEN T, GUO X, et al. The role of oxidative stress and inflammation in X-link adrenoleukodystrophy[J]. Front Nutr, 2022,9:864358.
[20]LAN F H, WANG Z H, KE L F, et al. A rapid and sensitive protocol for prenatal molecular diagnosis of X-linked adrenoleukodystrophy[J]. Clin Chim Acta, 2010,411(23-24):1992-1997.
[21]EICHLER F, DUNCAN C, MUSOLINO P L, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy[J]. N Engl J Med, 2017,377(17):1630-1638.
[22]CARTIER N, HACEIN-BEY-ABINA S, BARTHOLOMAE C C, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy[J]. Science, 2009,326(5954):818-823.
[23]MOSER H W, RAYMOND G V, LU S E, et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzos oil[J]. Arch Neurol, 2005,62(7):1073-1080.
[24]TIEU J H, SAHASRABUDHE S A, ORCHARD P J, et al. Translational and clinical pharmacology considerations in drug repurposing for X-linked adrenoleukodystrophy——A rare pe-roxisomal disorder[J]. Br J Clin Pharmacol, 2022,88(6):2552-2563.
[25]MONTERNIER P A, SINGH J, PARASAR P, et al. Therapeutic potential of deuterium-stabilized (R)-pioglitazone-PXL065-For X-linked adrenoleukodystrophy[J]. J Inherit Metab Dis, 2022,45(4):832-847.
(本文編辑 范睿心 厉建强)
