基于噁三唑设计潜在高能量密度化合物
2023-04-04荆苏明王媛媛苗舒月邓佳豪
朱 锦,荆苏明,李 聪,王媛媛,苗舒月,邓佳豪
(中北大学 环境与安全工程学院,山西 太原 030051)
引 言
近年来,高氮含能化合物已成为各国的研究热点。高氮含能化合物普遍具有高能量密度、高的正生成热、易于实现氧平衡、低污染性以及释放能量过程中低信号特征等诸多优点[1-3],被用作高能量密度材料以提升武器弹药的作战能力。
在研究高氮化合物的过程中发现了一种新型含能化合物结构——噁三唑。噁三唑具有绿色富氮高能结构,氮含量为60%,所有原子处于一个平面,具有良好的平面性,其分子内具有潜在硝基和潜在叠氮基结构,这些对含能化合物的密度、生成热、氧平衡、爆速和爆压有良好的促进作用[4-8];Matthew L. Gettings等[9-11]分别在2020年和2021年报道了新型含能化合物3-(1,2,3,4-四唑)-1,2,3,4-噁三唑-5-氧化物和3-(3-硝基-1,2,4-三唑)-1,2,3,4-噁三唑-5-氧化物,其理论密度均在1.85g/cm3左右,爆速分别为8906m/s和8360m/s,爆压均在30GPa左右。Peter Politzer等[12]模拟计算结果表明,以噁三唑为框架设计的高能量密度化合物是非常有潜力的,如1,2,3,5-噁三唑-3-氧化物,密度1.77g/cm3,爆速8.93km/s,爆压35.0GPa,经静电势分析该化合物具有较低的感度。总之,噁三唑具有良好的开发价值,有望成为高能量密度化合物的结构单元。
基于此,本研究以1,2,3,4-噁三唑和1,2,3,5-噁三唑作为基本构建单元,设计了A、B两组噁三唑类含能化合物,并且以1,2,4-三唑设计的C组化合物作为对比噁三唑类化合物能量性能的对照组。利用量子化学理论对噁三唑类化合物的性能(密度、生成焓、爆轰参数、稳定性等)进行研究,从中挑选出性能优异的分子结构,为后续噁三唑类含能化合物的研究工作提供理论依据。
1 计算方法
1.1 分子构建
本研究在两种噁三唑和1,2,4-三唑构建单元的基础上引入了—NO2、—NHNO2、—C(NO2)3、—CF(NO2)2、—CNF2(NO2)2五种含能基团,设计了A、B两组噁三唑类化合物和C组对照化合物,如图1所示。运用Gaussian 09W程序[13],通过密度泛函[14]方法B3PW91/6-31G(d,p)基组优化设计的四组分子构型,对优化后的结构进行频率振动分析。
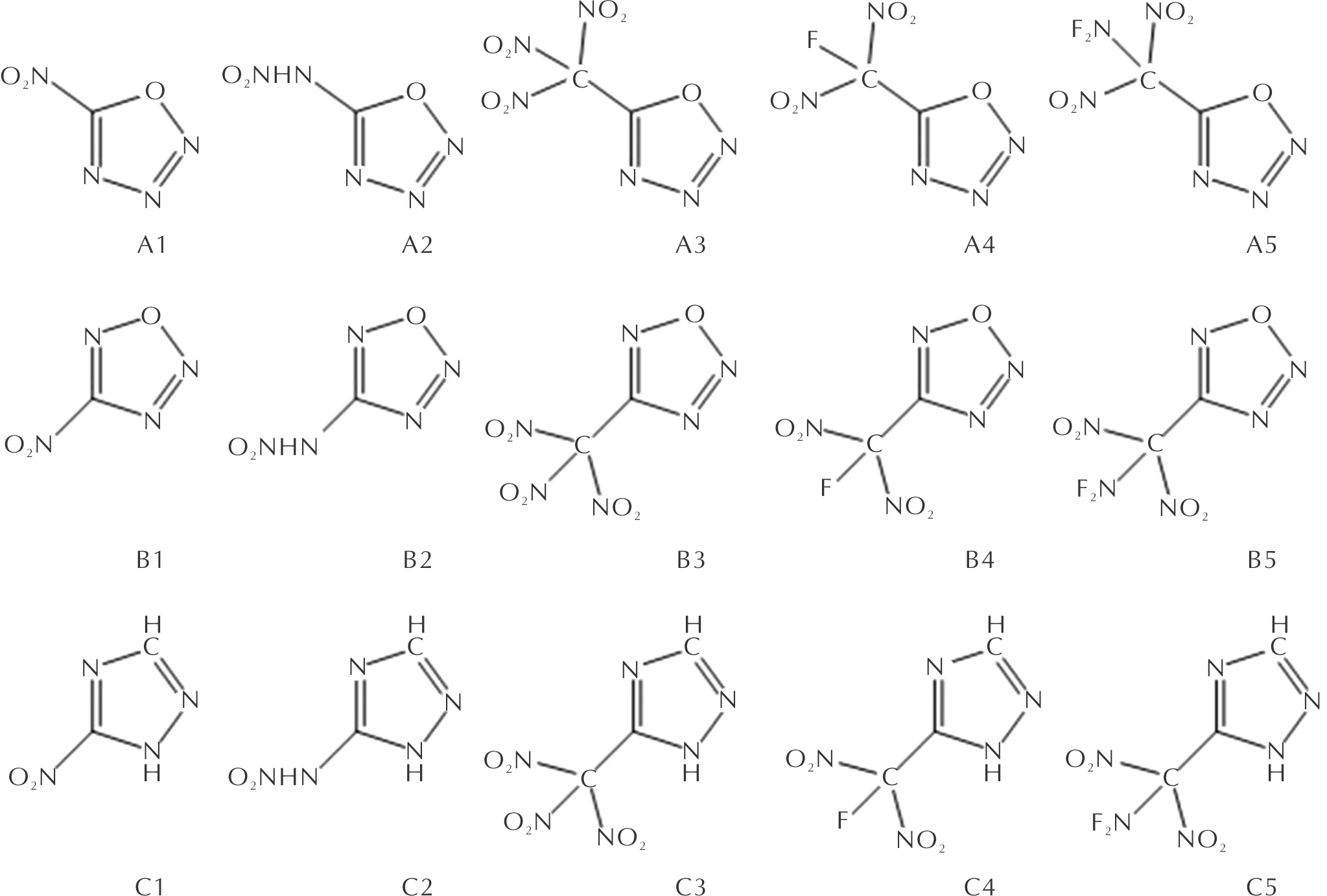
图1 A、B、C三组化合物的分子结构Fig.1 Molecular structure of compounds A, B and C
1.2 理论密度
在优化构型基础上,采用Montecarlo方法[15]得到化合物分子的理论摩尔体积(Vm,cm3/mol),结合Rice等[16]提出的公式预测含能化合物的理论密度,见式(1):
(1)
式中:M为化合物分子的摩尔质量,g/mol;Vm为分子体积,cm3/mol;υ为静电平衡系数;σ2为静电势总方差,kcal2/mol2。
1.2 生成焓
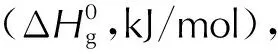

图2 噁三唑类化合物等键反应方程式Fig.2 Isobond reaction equations of oxatriazole compounds
ΔH298=∑ΔHf,P-∑ΔHf,P
(2)
ΔSH0=0.000267S2+2.966087+1.650087(υσ2)0.5
(3)
(4)
式中:P为生成物;R为反应物;ΔHf,P为298.15K下产物的生成焓,kJ/mol;ΔHf,R为298.15K下反应物的生成焓,kJ/mol;S为分子表面静电势总面积,Å2。
1.3 爆轰参数
本研究根据含能化合物的固相生成焓、密度和分子组成,采用Kamlet-Jacobs公式[19]计算化合物的爆速(D,km/s)和爆压(P,GPa),见式(5)和式(6):
(5)
(6)
式中:P为炸药装药密度,g/cm3;N为每克炸药生成气体产物的总摩尔数,mol/g;M为爆炸气体产物的平均分子量,g/mol;Q为化合物的爆热值,cal/g。
1.4 热稳定性
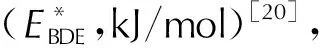
EBDE=E(A·)+E(B·)-E(A-B)
(7)
(8)
式中:E(A·)和E(B·)分别为化合物断裂后的能量,kJ/mol;E(A-B)为目标化合物的总能量,kJ/mol。
2 结果与讨论
2.1 噁三唑分析
经过Gaussian 09W程序的优化,1,2,3,4-噁三唑和1,2,3,5-噁三唑的模型如图3所示,1,2,3,4-噁三唑的N—N、C—O、N—O的键长分别为1.37、1.32、1.42Å;1,2,3,5-噁三唑的C—N、N—O、N—O的键长分别为1.37、1.34Å、1.38Å,这些键的键长均处于其典型的单键和双键之间,表明两个噁三唑环均具有共轭体系。
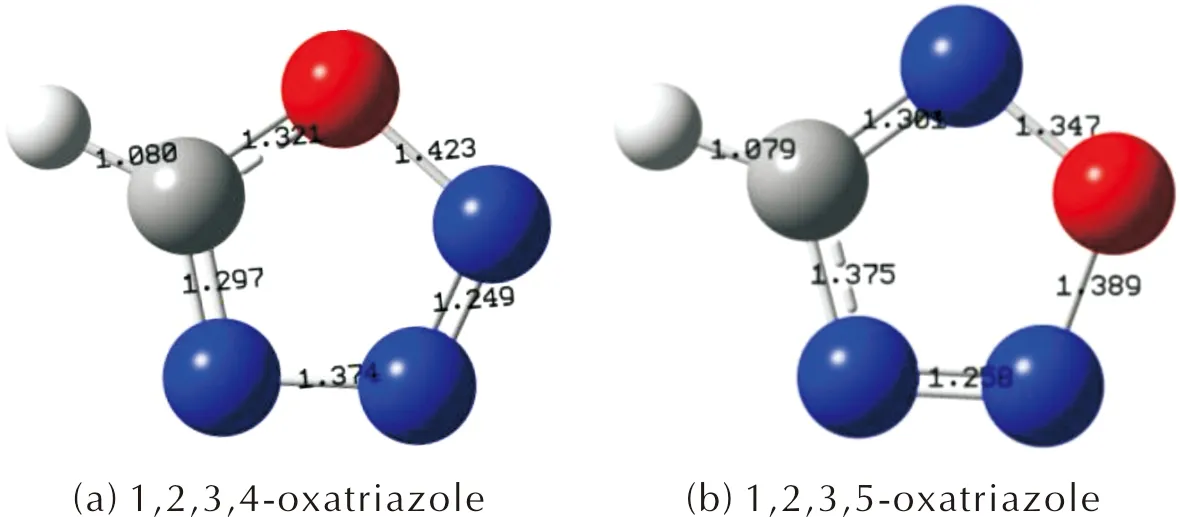
图3 1,2,3,4-噁三唑和1,2,3,5-噁三唑优化模型Fig.3 Optimization models of 1,2,3,4-oxatriazole and 1,2,3,5-oxatriazole
使用Multiwfn软件对1,2,3,4-噁三唑和1,2,3,5-噁三唑的ELF分析并绘制分析图,结果如图4所示。两个噁三唑骨架中的N—N、C—O、N—O、C—N的之间高定域区域具有较高的ELF值,表现出了共价键性质。上述表明噁三唑骨架具有一定的共轭性[21]。
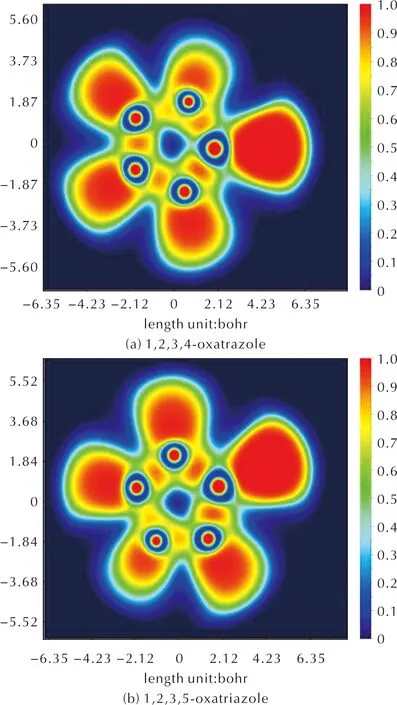
图4 1,2,3,4-噁三唑和1,2,3,5-噁三唑ELF投影图Fig.4 ELF projections of 1,2,3,4-oxatrazole and 1,2,3,5-oxatrazole
2.2 目标化合物几何构型分析
经过Gaussian 09W程序的优化,3组化合物分子的几何构型如图5所示。3组化合物分子的优化结果中均无虚频出现,表明优化结果可靠,A、B两组噁三唑类的部分化合物键长和Mayer键级的计算结果见表1。
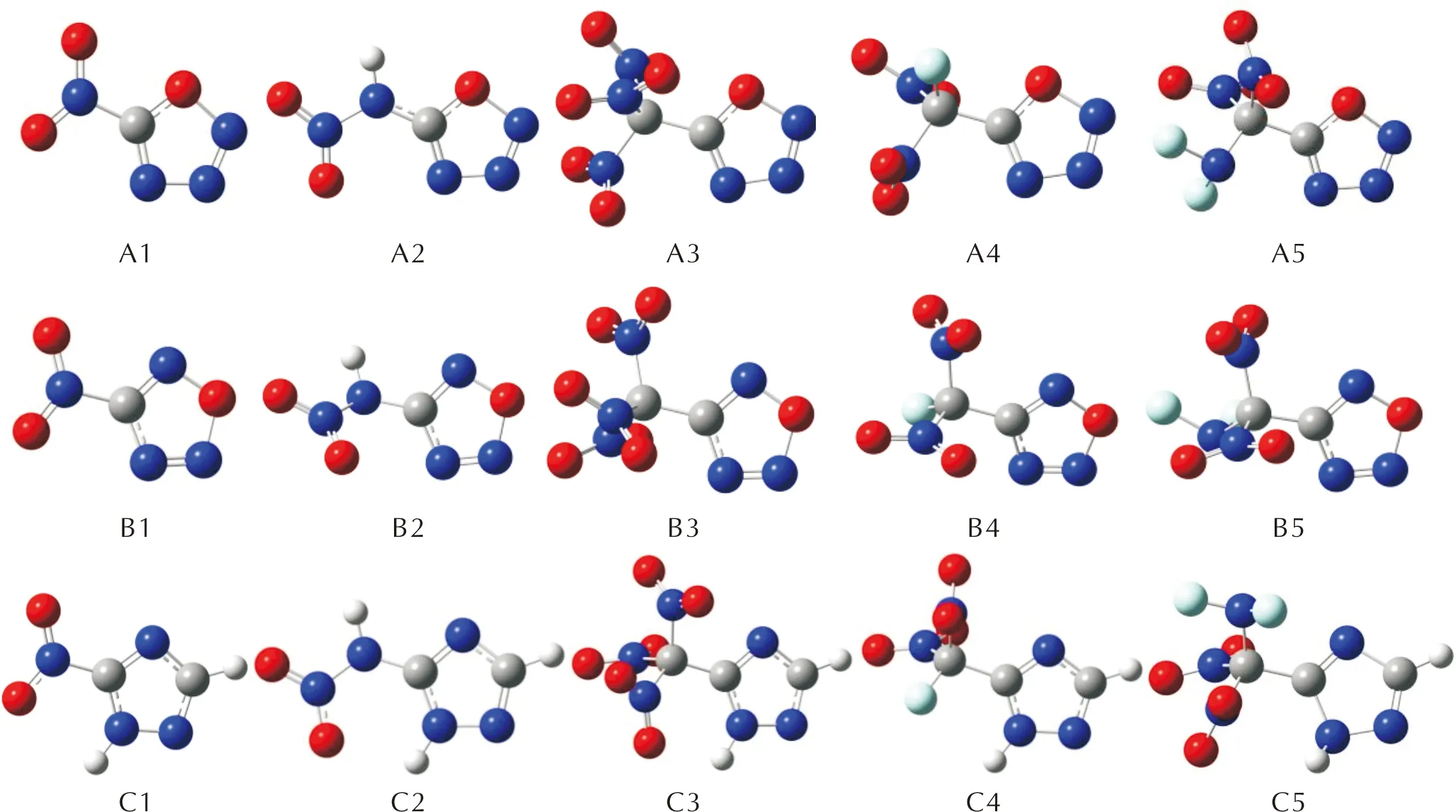
图5 目标化合物分子的几何构型Fig.5 Geometric configurations of target compounds
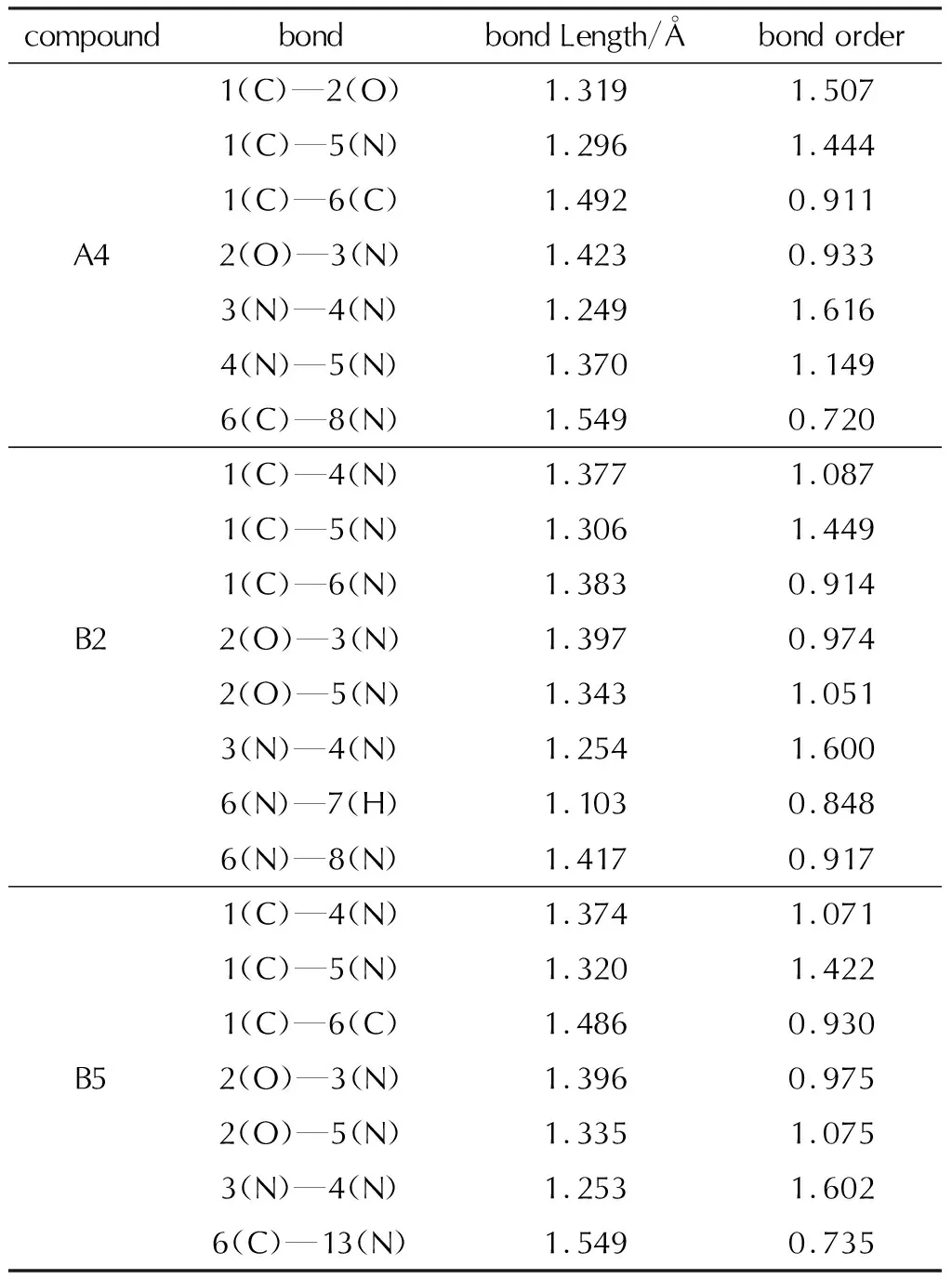
表1 化合物的部分键长和Mayer键级Table 1 Partial bond lengths and Mayer bond orders of compounds
通过对A、B两组键长的分析,A、B两组化合物中噁三唑骨架的N—N、C—O、N—O、C—N的键长分别处于其典型单键和双键之间,表明噁三唑在含能基团作用下仍具有一定的共轭效应;在基团—CF(NO2)2和—CNF2(NO2)2中C—N(NO2)键长均在1.54Å以上,Mayer键级均处于0.73左右,这两个原子间的电子云密度重叠较小,表明该键可能会在外界作用下优先断裂,这可能成为分子稳定性的短板。
2.3 性能分析
在相同基组下对A、B两组噁三唑类化合物和对照组C组的三唑类化合物的性能参数(密度、固相生成焓、爆轰参数)进行计算,结果如表2所示。
从表2可以看出,A、B两组化合物相比于C组表现出更高的密度,这可能是由于噁三唑中的氧原子体积小,导致原子间排布更紧凑,从而使噁三唑类理论密度大于三唑,这一结果表明,噁三唑相较于三唑是更具有潜力的高能量密度化合物构建框架;分析A、B两组化合物可发现,1,2,3,4-噁三唑和1,2,3,5-噁三唑的理论密度几乎一致,这一现象说明噁三唑的两个同分异构体并不会对密度产生很大影响;通过对A组的5种化合物对比发现,A3、A4、A5的密度明显高于A1、A2,表明含能基团—C(NO2)3、—CF(NO2)2以及—CNF2(NO2)2能够显著提高化合物密度,对比B组和C组也可得到相同结论。基团与密度的大小关系为:
—CNF2(NO2)2>—CF(NO2)2>—C(NO2)3>—NHNO2>—NO2
固相生成焓是影响高能量密度化合物爆速、爆压的因素之一。由表2可知,A、B两组的生成焓大于C组,B组的生成焓大于A组,大部分化合物具有较高的正生成焓,其中B3的生成焓最高,为237.09kJ/mol。含有—CF(NO2)2和—CNF2(NO2)2的化合物都具有负的生成焓。在噁三唑骨架相同时,含能基团与生成焓的大小关系为:

表2 化合物的爆轰参数计算结果Table 2 Calculation results of detonation parameters of compounds
—C(NO2)3>—NHNO2>—NO2>—CF(NO2)2>—CNF2(NO2)2
A、B两组氧平衡介于6.11%~23.89%之间,均为正氧平衡,C组均为负氧平衡。分析数据发现,噁三唑中潜在的硝基结构可显著改善化合物的氧平衡,官能团中硝基和氟对化合物氧平衡也有很大的提升。
通过表2可以发现,A、B两组化合物的爆轰性能相近,均超过C组对应的三唑化合物,其中,A4、A5、B4、B5的爆速超过了9.0km/s,A4、B4、A5、B5的爆压均超过40GPa,达到了预期高能量密度化合物的定量要求(D>9.0km/s,P>40GPa);该结果表明噁三唑类化合物的爆轰性能优于三唑,在构建高能量密度化合物方面比三唑更具有潜力。分析官能团发现,引入—C(NO2)3、—CF(NO2)2和—CNF2(NO2)2能够显著提升化合物的爆速和爆压,—NHNO2和—NO2的提升幅度较小。对比化合物A3、A4、A5,发现3个官能团对化合物能量的贡献顺序为:—CNF2(NO2)2>—C(NO2)3>—CF(NO2)2,分析B组和C组也会发现同样的规律。由此可见,在两种噁三唑骨架上引入—C(NO2)3、—CF(NO2)2和—CNF2(NO2)2是设计高能量密度化合物的有效途径。
2.4 前线轨道能量分析
轨道能量差与分子的化学稳定性、化学反应活性有关[22-23],在B3PW91/6-31G(d,p)水平下计算含能化合物分子轨道能量差(ΔELUMO-HOMO),计算结果如表3所示。

表3 化合物前线轨道能级差Table 3 Frontier orbital energy level difference of compounds
分析数据可知,A、B两组化合物的能隙大于C组对应化合物,B组的能隙大于A组,该结果表明目标化合物中噁三唑类比三唑类化学稳定性更好,其中,1,2,3,5-噁三唑类在3种目标化合物中最优。通过对A组(或B组)的目标化合物对比发现,A4(或B4)能隙最大,电子最不容易发生跃迁,预示其反应活性最低,化学稳定性最好;A1(或B1)的能隙最小,故化学稳定性最差,但C组无法得到此类结论,这可能是因为噁三唑对化学稳定性的影响所导致的。
2.5 热稳定性分析

表4 化合物最弱键级和键解离能Table 4 The weakest bond orders and bond dissociation energies of compounds
2.6 分子表面静电势和撞击感度分析
静电势与感度具有一定的相关性[24-25],利用Multiwfn程序和VMD程序绘制了A、B、C三组化合物在0.01a.u电子密度等值面上的静电势分布图,如图7所示,并且对三组目标化合物的正负静电区域面积强度进行统计,见表5。
由图7和表5可知,目标化合物的负静电势主要分布在骨架的氮原子、氧原子以及硝基氧和氟原子上;正静电势主要分布在中心环、硝铵基氢以及碳原子上;噁三唑和三唑骨架引入含氟和硝基的含能基团导致正静电势居中负静电势在边缘;3组化合物的正负电荷面积比(As+/As-)在0.89~1.47之间,A、B两组的正负电荷面积比明显大于C组,表明噁三唑类化合物与三唑类化合物相比具有更高的感度。通过表中数据对官能团分析发现,含有官能团—CNF2(NO2)2和—C(NO2)3的化合物感度高。根据判断化合物感度的方法(As+/As-接近1.5时感度较高)[19],A5、B5的感度在目标化合物中较高,化合物中A1和B1的感度较低。
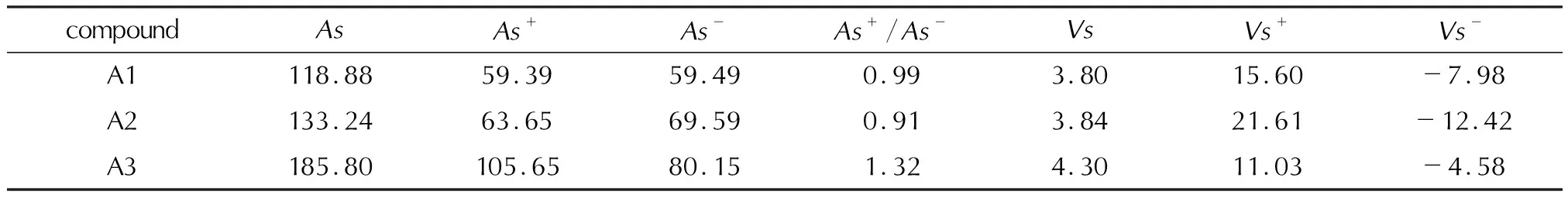
表5 化合物静电势参数Table 5 Electrostatic potential parameters of compounds
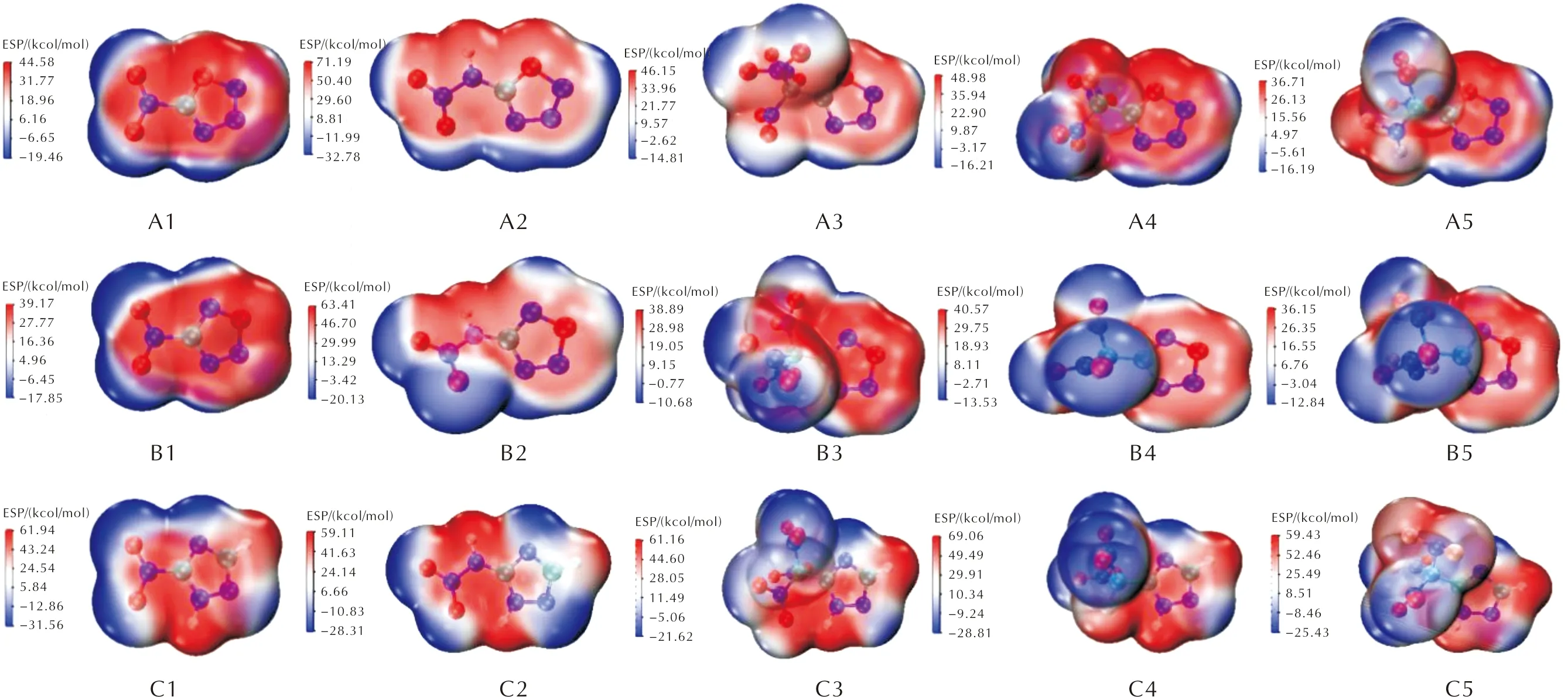
图7 噁三唑类化合物的静电势图Fig.7 Electrostatic potential diagrams of oxatriazole compounds
2.7 目标分子优选
根据上述分析结果,结合高能量密度化合物的基本要求,筛选出A3、A4、A5、B3、B4、B5潜在的高能量密度化合物分子见表6。

Continued

表6 目标分子优选Table 6 Target molecule optimization
3 结 论
(1)以噁三唑构建的含能化合物爆轰性能和化学稳定性优于三唑构建的含能化合物,热稳定性和感度比三唑类化合物差。
(2)引入的含能基团—CNF2(NO2)2、—CF(NO2)2、—C(NO2)3、—NHNO2、—NO2对于爆轰性能的贡献顺序为:—CNF2(NO2)2>—CF(NO2)2>—C(NO2)3>—NHNO2>—NO2,其中—CNF2(NO2)2、—CF(NO2)2、—C(NO2)3贡献的能量较大,是具有构建高能量密度化合物潜力的含能基团。
(3)含能基团—CNF2(NO2)2和—C(NO2)3的化学稳定性、热稳定性以及感度较差,相比于引入的其他基团的安全性差。
(4)计算结果表明,化合物A3、A4、A5、B3、B4、B5密度均在1.90g/cm3以上,爆速均在9.0km/s以上,爆压均在40GPa左右,达到了高能量密度化合物的要求,有望成为性能优良的高能量密度化合物。
(5)综合考量含能化合物的爆轰性能和安全性能,在高能量密度领域,噁三唑具有很大潜力,噁三唑结构为后续高能量密度化合物的设计与合成研究提供了新的思路。
