桑菊感冒颗粒质量标准完善研究
2023-04-01董秋香张月寒刘翀王腾姚辉桑皖怡付萍萍保定市食品药品检验所河北保定07000中国医科大学中英联合学院沈阳0000
董秋香,张月寒,刘翀,王腾,姚辉,桑皖怡,付萍萍*(. 保定市食品药品检验所,河北 保定07000;. 中国医科大学中英联合学院,沈阳 0000)
桑菊感冒颗粒源自清代医学家吴瑭所著《温病条辨》,收载于《卫生部药品标准·中药成方制剂(第二册)》,本方由桑叶、菊花、连翘、薄荷、苦杏仁、桔梗、甘草和芦根8味药材制成,方中桑叶、菊花为君药,薄荷、杏仁、桔梗为臣药,连翘、芦根为佐药,甘草为使药,调和诸药[1],共奏疏风清热、宣肺止咳之功效,传统用于风热感冒初起,头痛,咳嗽,2018年流行性感冒诊疗方案的轻症辨证治疗方案亦新增了桑菊感冒颗粒。经国家药品监督管理局官网检索桑菊感冒颗粒目前共有44个批准文号,涉及44家生产企业,现行质量标准仅收载了性状和颗粒剂通用检查项目[2],缺少专属性鉴别及量化指标,质量标准不完善,不能有效评价其内在质量,难以保证人们用药的安全有效。药品质量风险排查处置机制是我国首创的将抽检探索性研究结果与监督检查相结合的监管策略,是对药品监管科学理论的进一步丰富和发展[3]。本课题参考相关文献[4-6]开展探索性研究,建立薄层色谱(TLC)法鉴别菊花、连翘及甘草,建立高效液相色谱(HPLC)法同时测定连翘酯苷A、连翘苷及甘草酸的含量,对市售23批次样品进行综合分析,考察市售桑菊感冒颗粒的质量现状,为其建立完善的质量标准和提升整体质量水平提供技术支撑。
1 仪器与试药
1.1 仪器
LC-20AB 型高效液相色谱仪(配备PDA检测器,LCMS solution Ver3工作站,日本岛津公司);BP211D(德国赛多利斯公司,十万分之一);CAMAG 薄层色谱成像系统(瑞士CAMAG公司);SK250HP超声波清洗器(上海科导超声仪器有限公司);WGL-45B电热鼓风干燥箱(天津泰斯特仪器有限公司)。
1.2 试药
桑叶对照药材(批号:121123-200603)、菊花对照药材(批号:121384-201805)、连翘对照药材(批号:120908-201216)、薄荷对照药材(批号:120916-201812)、甘草对照药材(批号:120904-202021)、苦杏仁对照药材(批号:121554-201204)、桔梗对照药材(批号:121028-201612)、芦根对照药材(批号:121107-201706)、连翘酯苷A对照品(批号:111810-201707;纯度:97.2%)、连翘苷对照品(批号:110821-201816;纯度:95.1%)、甘草酸铵对照品(批号:110731-202021;纯度:96.2%)(中国食品药品检定研究院);硅胶G板、乙腈、甲醇为色谱纯(德国Merck公司),水为娃哈哈纯净水,其余试剂为分析纯;桑菊感冒颗粒(市售,A企业,批号:04190108,编号:A1;B企业,批号:180601、200506、210405,编号:B1~3;C企业,批号:191004、201004、211001、210405,编号:C1~4;D企业,批号:202011031、202101002、202012005、202011016,编号:D1~4;E企业,批号:201101、210402,编号:E1~2;F企业,批号:210103、201204、211204,编号:F1~3;G企业,批号:2012001、2108002、2110002,编号:G1~3;H企业,批号:21030002、21040005,编号:H1~2;I企业,批号:210603,编号:I1)。
2 方法与结果
2.1 样品情况
本次共收集样品23批次,涉及生产企业9家,占生产桑菊感冒颗粒全部企业的20%;均来自经营企业;规格均为11 g;贮藏条件均为密封;不同企业生产的样品有效期不一致,企业A、C、E、F生产的10批样品为24个月,企业B、D、G、H、I生产的13批样品为36个月。
2.2 法定标准检验
本品现行质量标准为《卫生部药品标准·中药成方制剂(第二册)》,检验项目包括性状、粒度、水分、溶化性、装量差异、微生物限度,对23批样品依据现行标准进行全项检验,合格率为100%。
2.3 TLC 鉴别
2.3.1 菊花 取本品2 g,研细,加水20 mL,振摇使溶解(必要时过滤),用水饱和的正丁醇振摇提取2 次,每次20 mL,合并正丁醇提取液,用正丁醇饱和的水20 mL洗涤,蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液;取处方中各药味对照药材,以模拟处方比例及方法制备阳性样品,同法制成阳性样品溶液(编号:Y1)。取菊花对照药材0.1 g,加水20 mL加热回流1 h,滤过,同法制成对照药材溶液。以模拟处方方法制备缺菊花的阴性样品,同法制成阴性对照溶液。固定相为硅胶G,展开剂为甲苯-乙酸乙酯-甲酸-冰醋酸-水(1∶15∶1∶1∶2)的上层溶液;点样量为5 μL;展开,晾干,喷以2%三氯化铝乙醇溶液,加热至干,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与菊花对照药材溶液色谱相应位置上显相同颜色的荧光斑点,且阴性样品无干扰。色谱图见图1。
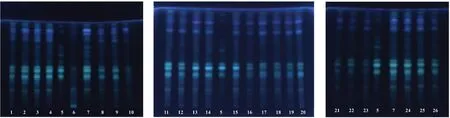
图1 菊花薄层色谱图Fig 1 TLC chromatogram of Chrysanthemi Flos
2.3.2 连翘 供试品溶液同“2.3.1”项下方法制备。取连翘对照药材0.2 g,加水20 mL加热回流1 h,滤过,同法制成对照药材溶液。取连翘苷对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。以模拟处方方法制备缺连翘的阴性样品,同法制成阴性样品溶液。固定相为硅胶G,展开剂为三氯甲烷-甲醇-甲酸(9∶2∶0.1);点样量为10 μL;展开,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,日光下检视。结果供试品溶液色谱中,在与连翘对照药材溶液色谱相应位置上显相同颜色的斑点,且阴性样品无干扰。色谱图见图2。

图2 连翘薄层色谱图Fig 2 TLC chromatogram of Forsythiae Fructus
2.3.3 甘草 供试品溶液同“2.3.1”项下方法制备。取甘草对照药材0.1 g,加水20 mL加热回流1 h,滤过,同法制成对照药材溶液。以模拟处方比例及方法制备缺少甘草的阴性样品,同法制成阴性样品溶液。固定相为硅胶G,展开剂为三氯甲烷-甲醇-水(13∶6∶2)10℃以下放置的下层溶液;点样量为10 μL;展开,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,日光下检视。结果供试品溶液色谱中,在与甘草对照药材溶液色谱相应位置上显相同颜色的斑点,且阴性样品无干扰。色谱图见图3。
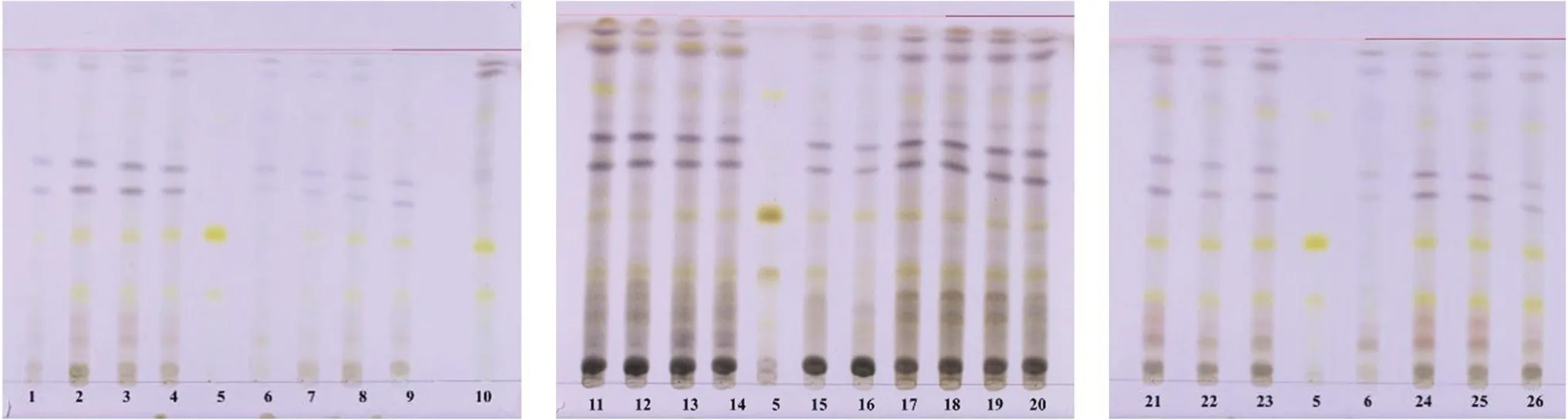
图3 甘草薄层色谱图Fig 3 TLC chromatogram of Glycyrrhizae Radix Et Rhizoma
2.4 多指标成分含量测定
2.4.1 色谱条件 色谱柱:Waters SunFire C18(4.6 mm×250 mm,5 μm),流动相:0.1%磷酸溶液(A)-乙腈(B),梯度洗脱(0~40 min,10%~18%B;40~55 min,18%~36%B;55~65 min,36%~60%B;65~70 min,60%~100%B;70~71 min,100%~10%B;71~85 min,10%B),流速:1.0 mL·min-1,柱温:30℃;检测波长:237 nm;进样量:10 μL。
2.4.2 溶液的制备 取连翘酯苷A、连翘苷、甘草酸铵对照品适量,精密称定,加80%甲醇制成质量浓度分别为42.85、14.17、14.56 μg·mL-1的混合溶液,作为对照品溶液。取本品10袋,内容物研细,取细粉1 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25 mL,称定重量,超声处理(功率:250 W,频率:53 kHz)30 min,放冷,用80%甲醇补足减失重量,摇匀,滤过,取续滤液作为供试品溶液。分别取处方中各药味对照药材,以模拟处方比例及方法制备缺少连翘、甘草的阴性样品,照供试品溶液的制备方法,同法制成缺连翘的阴性样品溶液、缺甘草的阴性样品溶液。
2.4.3 方法学考察
① 专属性试验:取对照品溶液、供试品溶液、阴性样品溶液及空白溶剂,进样测定,记录色谱图(见图4),供试品溶液色谱图中峰1与相邻峰的分离度分别为1.75、1.71,峰2与相邻峰的分离度分别为3.94、2.83,峰3与相邻峰的分离度分别为5.38、2.68,均大于1.5。同时采用PDA检测器在190~800 nm进行光谱扫描,峰1、2、3的紫外光谱图分别与连翘酯苷A、连翘苷、甘草酸对照品的紫外光谱图一致。峰1、2、3的峰纯度检测结果显示未检测到杂质,依据工作站判定规则可判断为单一成分。结果表明,此色谱条件下3种待测成分峰与相邻峰分离良好,阴性样品无干扰。

图4 高效液相色谱图Fig 4 HPLC chromatograms
② 线性关系考察:取连翘酯苷A、连翘苷、甘草酸铵对照品适量,精密称定,加80%甲醇定量稀释制成质量浓度分别为96.71、76.08、75.87 μg·mL-1的混合对照品储备液,加80%甲醇等倍逐级稀释成系列浓度的对照品溶液,进样测定,记录峰面积。分别以3种待测成分的质量浓度(X,μg·mL-1)为横坐标,峰面积(Y)为纵坐标进行线性回归,结果各待测成分在相应范围内线性关系良好,回归方程及线性范围见表1。

表1 主成分回归方程、相关系数及线性范围Tab 1 Regression equation,correlation coefficient and linear range of each component
③ 定量限和检测限:取“2.4.3”项下的对照品储备液适量,等倍逐级稀释,进样测定,结果连翘酯苷A、连翘苷及甘草酸的定量限(LOQ)分别为0.47、0.23、0.45 μg·mL-1(S/N=10);检测限(LOD)分别为0.14、0.07、0.14 μg·mL-1(S/N=3)。
④ 精密度试验:取“2.4.2”项下对照品溶液,进样测定,记录峰面积。结果连翘酯苷A、连翘苷、甘草酸峰面积的RSD分别为0.20%、0.40%、0.30%(n=6),表明仪器精密度良好。
⑤ 重复性试验:取同一批样品(编号B1),按“2.4.2”项下方法制备供试品溶液6份,进样测定,以外标法计算平均含量。结果样品中连翘酯苷A、连翘苷及甘草酸的平均含量分别为1.087、0.3827、0.4485 mg·g-1,RSD分别为0.60%、0.60%、0.70%(n=6),表明本方法重复性良好。
⑥ 稳定性试验:取“2.4.2”项下供试品溶液(编号:B1),分别于室温下放置0、1.5、3、4.5、6、7.5、9、12、18、24 h,进样测定,记录峰面积。结果连翘酯苷A、连翘苷及甘草酸色谱峰面积的RSD分别为0.20%、0.40%、0.40%(n=10),表明供试品溶液在室温下24 h内稳定。
⑦ 加样回收试验:取已知含量的样品(编号B2)0.5 g,精密称定(9份),置100 mL具塞锥形瓶中,分别精密加入连翘酯苷A、连翘苷、甘草酸铵对照品适量,依法制备高、中、低浓度的供试品溶液,进样测定,计算加样回收率。结果3种成分的回收率均在97.57%~103.06%,RSD均小于2.0%。
2.4.4 含量测定 取23批样品,按“2.4.2”项下方法制备供试品溶液,进样测定,记录峰面积,以外标法计算3种成分的含量(甘草酸的计算结果与0.9797相乘),结果见表2,23批样品连翘酯苷A、连翘苷、甘草酸的平均含量为4.68、1.86、3.60 mg/袋,RSD值为72.1%、58.7%、44.1%。
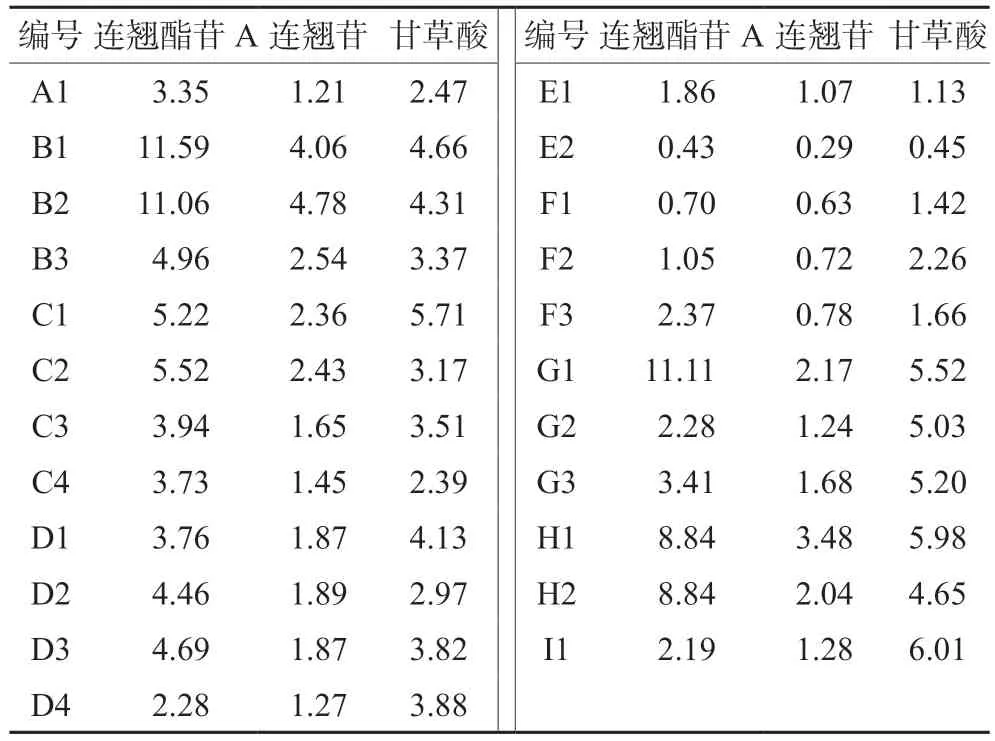
表2 样品含量测定结果(mg/袋)Tab 2 Content determination in the samples (mg/袋)
3 讨论
3.1 指标成分的确立
本研究曾尝试开展处方中君药桑叶的TLC鉴别,但选用多个展开系统展开后,均未排除阴性样品干扰,桑叶的鉴别有待进一步研究。本品由8味药材组方,其中薄荷采用气相色谱法测定薄荷脑的含量,桔梗采用蒸发光散射检测器测定桔梗皂苷D的含量,芦根质量标准中未收载含量测定,文献报道经碱性乙醇水溶液回流提取后测定芦根中对香豆酸和阿魏酸的含量[7],故未实现上述3味药材的同时测定。王庆喜等[8]用盐酸溶液回流提取后测定桑菊感冒颗粒中槲皮素的含量,亦未实现槲皮素的同时测定。本品中绿原酸、芦丁、苦杏仁苷的含量较低,且绿原酸为桑叶和菊花的共有成分,芦丁为桑叶、菊花和连翘的共有成分,均不适宜做含量测定的指标成分。本课题组前期进行了指纹图谱相关研究,共标定21个共有峰,属桑叶、菊花、连翘、甘草和薄荷,运用SPSS 18.0 统计分析软件进行主成分分析,筛选出8个差异性指标成分,结合色谱峰的保留时间及紫外光谱信息进行解析指认出其中3个色谱峰,分别为连翘酯苷A、连翘苷和甘草酸,其中连翘酯苷A和连翘苷属连翘,甘草酸属甘草,故本研究确立此3种成分为含量测定的指标成分。
3.2 提取溶剂及方式的选择
TLC鉴别考察用水饱和的正丁醇、乙酸乙酯、三氯甲烷、乙醚提取样品,按拟定色谱条件测定,结果以水饱和的正丁醇为溶剂提取时,样品信息更丰富,斑点更清晰,故选择以水饱和的正丁醇作为提取溶剂;HPLC多指标成分含量测定考察提取溶剂(甲醇、90%甲醇、80%甲醇、水)和提取时间(10、20、30、40 min)对3种成分提取效率的影响,结果显示以80%甲醇超声30 min时3种成分的含量较高,故选择此方法制备供试品溶液。
3.3 色谱条件的优化
3.3.1 TLC色谱条件 曾尝试用相同色谱系统同时分析连翘和甘草,但在甘草的色谱系统下连翘苷的Rf值偏低,连翘的色谱系统下甘草主斑点的Rf值均偏高,最终确定本文的色谱条件分别测定连翘和甘草。
3.3.2 HPLC流动相 甲醇-水系统洗脱能力较差,无法检测到甘草酸峰,以乙腈-水为流动相时峰形较差,采用乙腈-0.05%磷酸溶液、乙腈-0.1%磷酸溶液和乙腈-0.2%磷酸溶液为流动相梯度洗脱,乙腈-0.05%磷酸溶液系统下色谱峰形较差,后两者差异不大,分离度符合要求且峰形较好,故选择乙腈-0.1%磷酸溶液为流动相。
3.4 含量测定限度制定
由于青翘与老翘中连翘酯苷A含量存在一定差异[9],而本品制法项下未明确青翘或老翘投料,且不同厂家生产工艺与设备不同造成的转移率差异,故未能采用样品的测定结果结合转移率确定含量限度。国家药品标准工作手册规定中药材和中药饮片原则上按照平均值的-20%制定含量测定限度,另2017年3月18日广东省药品检验所网站发布的关于“在广东省提高国家药品标准行动计划有关问题的解答”中提到,中成药含量限度一般按平均值下浮20% 较合适,故以23批次样品含量测定结果的平均值下浮20%为标准,将连翘酯苷A、连翘苷、甘草酸的限度暂定为3.74、1.49、2.88 mg/袋。针对青翘与老翘中连翘酯苷A含量的差异,有必要对本品中连翘酯苷A的含量制订更加科学的限度,此项工作有待进一步研究。
3.5 整体质量分析
23批样品按现行标准进行法定检验全部合格,但结合探索性研究结果深入分析,仍可发现一些与药品质量相关的问题。TLC鉴别结果分析23批样品均检出与菊花、甘草、连翘对照药材和连翘苷对照品相应的斑点,但斑点深浅不一;以本文制订的限度进行检验,有11批样品不合格,不合格率为47.8%,不合格样品涉及7家企业;23批样品的3种成分含量测定结果RSD值显示样品间质量存在显著差异,特别是连翘酯苷A的RSD值超过70%,推测可能是连翘采收期及生产工艺不同导致连翘酯苷A含量差异尤为显著。本文运用SPSS 18.0统计学软件对含量测定与水分、装量及粒度的相关性进行了分析,结果显示样品中连翘酯苷A和连翘苷的含量与水分成显著正相关,提示生产过程中干燥方式对含量测定结果有影响,建议生产企业严格控制干燥温度及时间。
3.6 有效期的制订
现行质量标准在关键检验项目上的缺失,致使有效期的制订不科学,且不同企业产品的有效期不同,建议生产企业对有效期重新考察,且不能忽略包装材料对有效期的影响。
桑菊感冒颗粒现行质量标准在项目设置上未关注影响药品有效性和安全性的重要质量要素,无法准确评价产品质量,标准亟待完善;法定标准检验合格率较高,探索性研究结果显示市售桑菊感冒颗粒质量存在显著差异,产品质量亟待提高。本文建立的TLC鉴别和HPLC含量测定方法填补了桑菊感冒颗粒关键质控项目的缺失,为完善质量标准和评价产品质量现状提供了重要依据。
