藏药长梗金腰中2个活性成分的提取富集工艺研究 Δ
2023-03-17黎运芬陈思尼珍向佳美慕泽泾朱玉野龚淑芳任刚江西中医药大学中药资源与民族药研究中心南昌0004西藏自治区食品药品检验研究院拉萨850000江西中医药大学附属医院国医堂南昌0004
黎运芬 ,陈思 ,尼珍 ,向佳美 ,慕泽泾 ,朱玉野 ,龚淑芳 ,任刚 (.江西中医药大学中药资源与民族药研究中心,南昌 0004;.西藏自治区食品药品检验研究院,拉萨 850000;.江西中医药大学附属医院国医堂,南昌 0004)
长梗金腰Chrysosplenium axillareMaxim.为虎耳草科金腰属多年生草本植物,主产于陕西、甘肃南部、青海东南部和新疆等地,生于海拔2 800~4 700 m的林下、灌丛间或石隙。据《中国藏药植物资源考订》记载,长梗金腰为特色藏药“亚吉玛”的基原植物之一[1]。长梗金腰以干燥全草入药,其味苦性凉,清胆热,祛胆病[2],用于治疗胆病引起的发烧头痛、胆囊疾患、急性黄疸型肝炎等症[3]。本课题组前期研究采用肝内胆汁淤积 (intrahe‐patic cholestasis,IC)模型小鼠验证了长梗金腰抗IC的功效,并从中分离得到了2个高度甲氧基化的黄酮醇衍生物——chrysosplenoside A(CA)和 chrysosplenoside I(CI)[4];并发现这2种成分对IC模型小鼠的肝损伤具有保护作用,且优于熊去氧胆酸(目前临床治疗IC的一线药物)。由此可见,CA、CI作为抗IC药物具有良好的开发前景。基于此,本研究初探长梗金腰中CA、CI的提取富集工艺,以期为CA、CI的药物开发提供物质基础。
1 材料
1.1 主要仪器
本研究所用主要仪器有EX1600型高效液相色谱(HPLC)仪(上海伍丰科学仪器有限公司)、CP‑214型电子天平(上海奥豪斯仪器有限公司)、KQ‑5200DB型超声波清洗器(宁波新芝生物科技股份有限公司)、BT25S型电子分析天平(北京赛多利斯仪器系统有限公司)、GOLD‑SIM型冷冻干燥机(美国Siemon公司)、R‑210型旋转蒸发仪(瑞士Buchi公司)。
1.2 药品与试剂
长梗金腰药材于2018年7月采集自四川省壤塘县中壤塘乡,经江西中医药大学中药资源与民族药研究中心钟国跃研究员鉴定为长梗金腰C.axillareMaxim.全草。CA、CI标准品为本实验室自制,纯度经HPLC检测均大于98%。HPD‑500型和D101型大孔吸附树脂购自沧州宝恩化工有限公司;Diaion HP‑20型大孔吸附树脂购自日本三菱化学公司;色谱级乙腈购自德国Merck公司;质谱级乙酸购自上海阿拉丁生化科技股份有限公司;其余试剂为实验室常用规格。
2 方法与结果
2.1 CA和CI含量测定方法的建立
2.1.1 供试品溶液的制备 取干燥长梗金腰全草适量,粉碎过三号筛,取0.5 g粉末,精密称定,置于150 mL具塞锥形瓶中,精密加入25 mL 50%甲醇,密塞,称定质量;超声(功率250 W,频率40 kHz)处理45 min,取出放冷,称定质量;以50%甲醇补足减失的质量,摇匀,过0.22 μm微孔滤膜,取续滤液,备用。
2.1.2 混合对照品溶液的制备 取CA、CI对照品适量,精密称定,加入甲醇制成质量浓度分别为0.420 0、0.360 0 mg/mL的混合对照品溶液,备用。
2.1.3 色谱条件 色谱柱为Inertsil ODS‑2(250 mm×4.6 mm,5 μm);流动相为0.1%乙酸溶液(A)‑乙腈(B),梯度洗脱(0~10 min,10%B→20%B;10~20 min,20%B→25%B;20~40 min,25%B→40%B;40~50 min,40%B→50%B;50~60 min,50%B→80%B;60~70 min,80%B→95%B);流速为0.8 mL/min;柱温为35 ℃;检测波长为254 nm;进样量为10 μL。
2.1.4 专属性考察 取“2.1.1”“2.1.2”项下供试品溶液和混合对照品溶液适量,按“2.1.3”项下色谱条件进行分析,记录色谱图。结果显示,供试品溶液和混合对照品溶液图谱中CA和CI的保留时间一致,且分离度均大于1.5。结果见图 1。
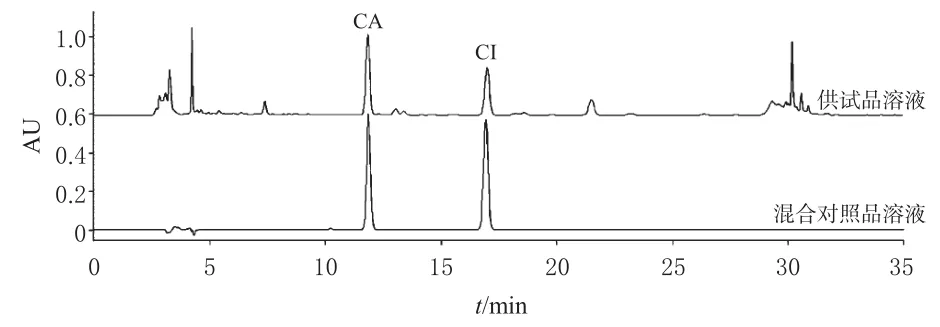
图1 供试品溶液和混合对照品溶液的HPLC图
2.1.5 线性关系考察 取“2.1.2”项下制备的混合对照品溶液适量,置于10 mL容量瓶中,用甲醇分别稀释2、4、8、16、32、64倍后得到系列对照品溶液;按“2.1.3”项下色谱条件进样分析,记录色谱图。以进样量(μg)为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得CA和CI的回归方程;再以信噪比10∶1、3∶1分别计算定量限和检测限。结果见表1。
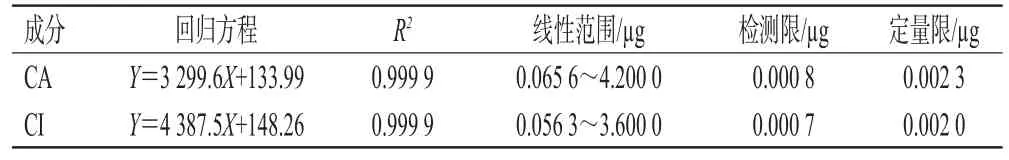
表1 CA和CI的线性关系及灵敏度考察结果
2.1.6 精密度试验 取同一供试品溶液适量,按“2.1.3”项下色谱条件重复测定6次,分别记录峰面积,计算得CA和CI峰面积的RSD分别为0.09%、1.36%(n=6),表明仪器精密度良好。
2.1.7 稳定性试验 取同一供试品溶液适量,分别于制备后0、2、4、8、12、24 h时按“2.1.3”项下色谱条件进样分析,记录峰面积,计算得到CA和CI峰面积的RSD分别为0.52%、0.82%(n=6),表明供试品溶液在制备后24 h内稳定性良好。
2.1.8 重复性试验 精密称取长梗金腰药材粉末约0.5 g,共6份,按“2.1.1”项下方法制备供试品溶液,再按“2.1.3”项下色谱条件进行分析,记录峰面积,并根据标准曲线分别计算CA和CI含量。结果显示,CA和CI含量的RSD分别为1.62%、1.04%(n=6),表明该方法重复性良好。
2.1.9 加样回收率试验 分别取CA、CI适量,精密称定,置于10 mL容量瓶中,加入甲醇溶解,制成CA、CI质量浓度分别为1.60、0.90 mg/mL的混合对照品溶液。精密称取适量已知含量的长梗金腰样品约0.25 g,平行6份,加入混合对照品溶液1 mL(相当于药材原有含量的100%);按“2.1.1”项下方法制备供试品溶液,再按“2.1.3”项下色谱条件进样分析,计算加样回收率。结果显示,CA、CI的平均加样回收率分别为95.96%、102.63%,RSD分别为1.88%、2.98%(n=6),表明该方法准确性良好。
2.2 长梗金腰中CA和CI的提取工艺优化
2.2.1 长梗金腰提取物的制备及CA和CI的总转移率测定 称取长梗金腰药材粉末1.0 g(过三号筛),置于150 mL具塞锥形瓶中,分别加入相应体积分数的乙醇溶液,密塞,静置12 h;于相应温度下超声(频率40 kHz,功率250 W,下同)提取45 min,过滤,滤液于40 ℃条件下减压回收乙醇,得浸膏状提取物。将所得浸膏状提取物置于-20 ℃条件下冷冻2 h,再置于冷冻真空干燥机中干燥,即得固态提取物。取固态提取物约0.1 g,精密称定,置于50 mL烧杯中,精密加入10 mL甲醇,超声使其充分溶解;取上清液过0.22 μm微孔滤膜,滤液按“2.1.3”项下色谱条件进样分析,根据标准曲线计算CA和CI的含量,再计算CA和CI的转移率(转移率=固态提取物中目标成分含量×提取物得率/生药材中目标成分含量×100%)。
2.2.2 正交实验 采用L9(34)正交实验对长梗金腰中CA和CI的提取工艺进行优化。以乙醇体积分数(A)、浸提温度(B)、浸提次数(C)及料液比(D)为考察因素,以CA和CI的总转移率为评价指标,根据L9(34)正交表设计实验,然后按“2.2.1”项下方法进行提取,计算CA和CI的总转移率。正交实验的因素与水平见表2,正交实验设计及结果见表3,方差分析结果见表4。
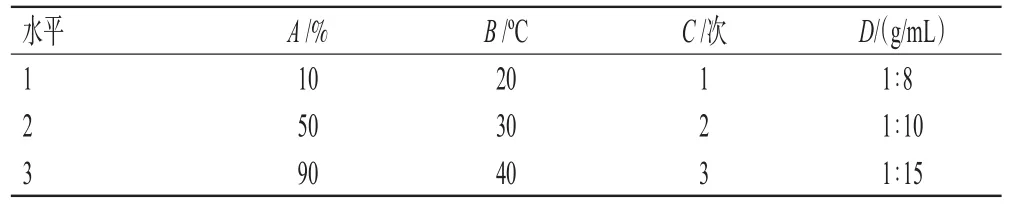
表2 正交实验的因素与水平
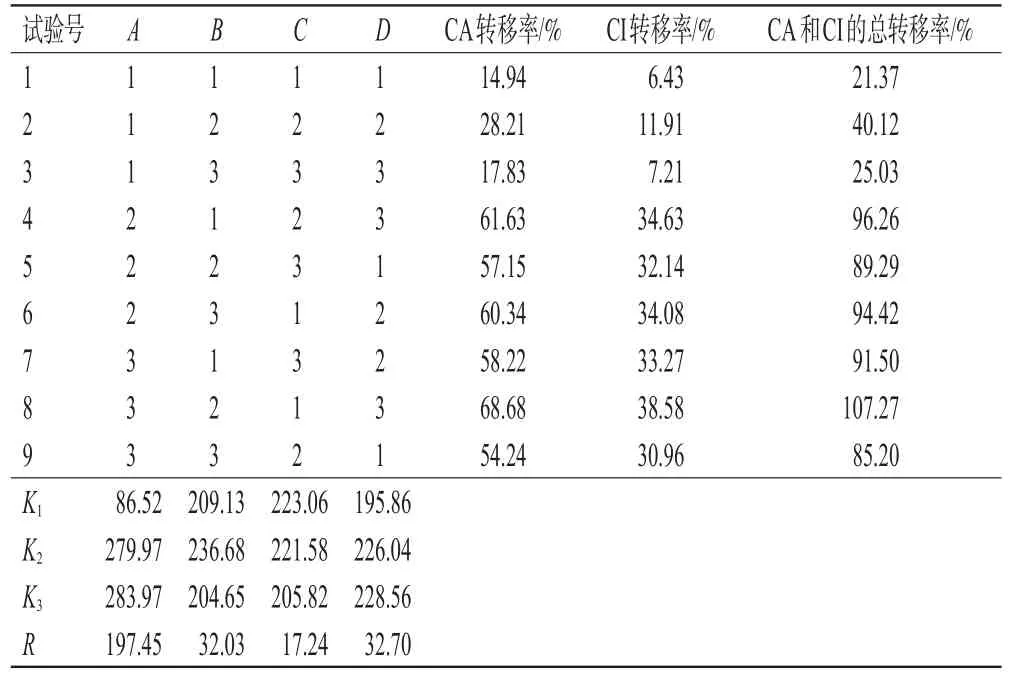
表3 正交实验设计及结果
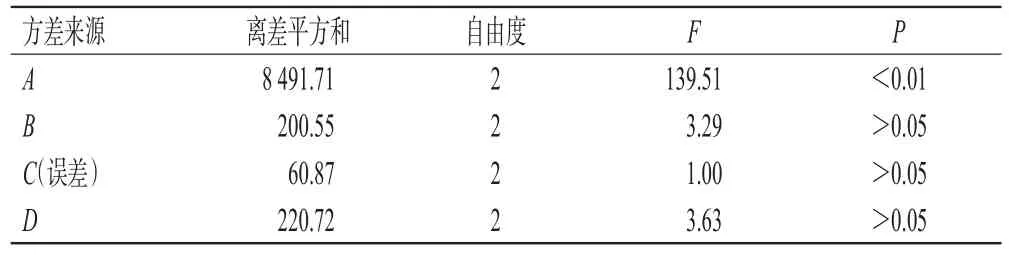
表4 方差分析结果
由表4可知,影响长梗金腰中CA和CI的总转移率的显著程度依次为A>D>B>C,即乙醇体积分数>料液比>浸提温度>浸提次数。其中,因素A对CA和CI的提取有显著影响(P<0.01),但其A2(50%乙醇)与A3(90%乙醇)水平的K值非常接近,从提取成本的角度考虑,本研究选择50%乙醇为提取溶剂。因素B、C、D对CA和CI的提取无显著影响,综合降低能耗、简化提取程序、节约溶剂的考虑分别选择室温、提取1次及料液比1∶8作为提取参数。因此,最终确定最优提取工艺为A2B1C1D1,即将长梗金腰药材粉末以8倍量的50%乙醇在室温下一次性超声提取45 min。
2.2.3 最优提取工艺的验证 按照“2.2.2”项下最优提取工艺条件提取长梗金腰中的CA和CI,平行3次。结果显示,长梗金腰中CA和CI的平均总转移率为95.43%,RSD为1.02%(n=3);平均总含量为25.55 mg/g,RSD为1.13%(n=3)。这表明该工艺合理、稳定、可行。
2.3 长梗金腰中CA和CI的富集工艺优化
笔者前期预实验考察了HP‑20、HPD‑500、D101型号大孔吸附树脂对长梗金腰中CA和CI总饱和吸附和洗脱的影响,结果发现,3种大孔吸附树脂对CA和CI的吸附效果均较好;但在洗脱过程中,相较于HP‑20、HPD‑500型大孔吸附树脂,D101型大孔吸附树脂吸附的CA和CI更易洗脱。因此,本研究笔者采用D101型大孔吸附树脂来富集长梗金腰中的CA和CI。由于柱层析过程中洗脱参数对CA和CI的富集具有重要的影响,因此笔者考察了杂质和目标成分的洗脱剂体积分数及用量对长梗金腰中CA和CI富集工艺的影响。
2.3.1 长梗金腰提取物的制备 取300.0 g长梗金腰按“2.2.2”项下的最优提取工艺提取长梗金腰中的CA和CI,得固态提取物,于4 ℃条件下保存备用。
2.3.2 上柱样品溶液的制备 取1.0 g长梗金腰固态提取物置于50 mL烧杯中,加入20倍量蒸馏水,搅拌均匀,超声处理10 min,过滤,即得质量浓度为50.0 mg/mL的样品溶液(以提取物计)。
2.3.3 洗脱剂体积分数的考察 称取1份处理好的D101型大孔吸附树脂66.0 g,湿法装柱,装入3 cm×35 cm的层析柱中[1柱床体积(BV)=75 mL]。将100 mL的样品溶液以2 BV/h的流速上柱,静置1 h,再依次用10%、20%、30%、40%、50%、60%、70%、80%和90%乙醇各洗脱4 BV,分别收集各段洗脱流分,采用HPLC法测定各样品洗脱流分中CA和CI的含量,并记录色谱图。结果如图 2所示,当乙醇体积分数<50%时,洗脱流分中CA和CI的总量(以mg计,下同)随着乙醇体积分数的升高而增大,且10%、20%乙醇洗脱流分中CA和CI的总量甚微;当乙醇体积分数>50%时,洗脱流分中CA和CI的总量呈下降趋势,且80%、90%乙醇洗脱流分中CA和CI的总量甚微。由此可知,50%乙醇可最大限度地将CA和CI解吸完全,故确定50%乙醇为CA和CI洗脱剂。20%乙醇洗脱流分中CA和CI的总量小,可将大部分极性较大的杂质洗脱下来,故确定20%乙醇为杂质洗脱剂。
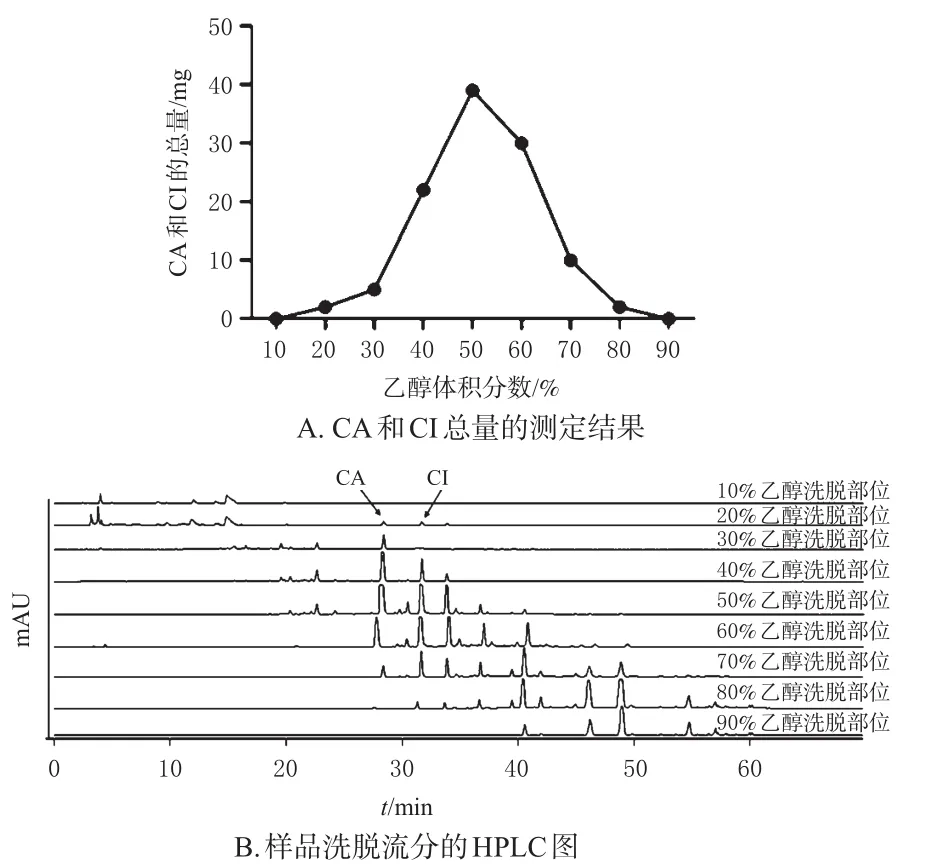
图2 洗脱剂体积分数对CA和CI解吸附效果的影响
2.3.4 杂质洗脱剂用量的考察 称取3份D101型大孔吸附树脂,每份66.0 g,湿法装柱,分别装入3 cm×35 cm的层析柱中。将100 mL样品溶液以2 BV/h流速上柱,静置1 h,分别以2、4、6 BV 20%乙醇洗脱杂质(流速为2 BV/h),然后用4 BV 50%乙醇以2 BV/h流速洗脱CA和CI,收集洗脱流分;采用HPLC法测定各样品洗脱流分中的CA和CI的含量,并记录色谱图。结果如图3所示,经2、4、6 BV 20%乙醇洗脱杂质后,进一步收集的4 BV 50%乙醇洗脱流分中CA和CI的总量分别为1.45、3.60、3.16 mg。由此可知,4 BV 20%乙醇洗脱杂质的效果较好。
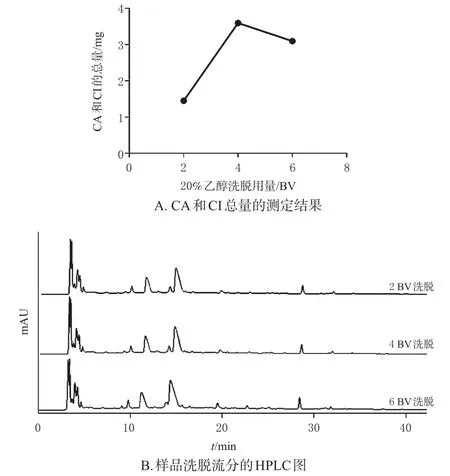
图3 杂质洗脱剂用量对杂质洗脱效果的影响
2.3.5 CA和CI洗脱剂用量的考察 称取1份D101型大孔吸附树脂66.0 g,湿法装柱,分别装入3 cm×35 cm的层析柱中。将100 mL样品溶液以2 BV/h流速上柱,静置1 h;用4 BV 20%乙醇冲洗杂质后,再用6 BV 50%的乙醇进行洗脱,每隔1 BV就收集一份洗脱流分;采用HPLC法测定各样品洗脱流分中CA和CI的含量,并记录色谱图。结果如图 4所示,第2 BV时50%乙醇洗脱流分中CA和CI的总量已达到最大值,第4 BV时50%乙醇洗脱流分中CA和CI的总量很低,说明到第4 BV时已基本洗脱完全。因此,确定4 BV为50%乙醇洗脱剂的最佳用量。
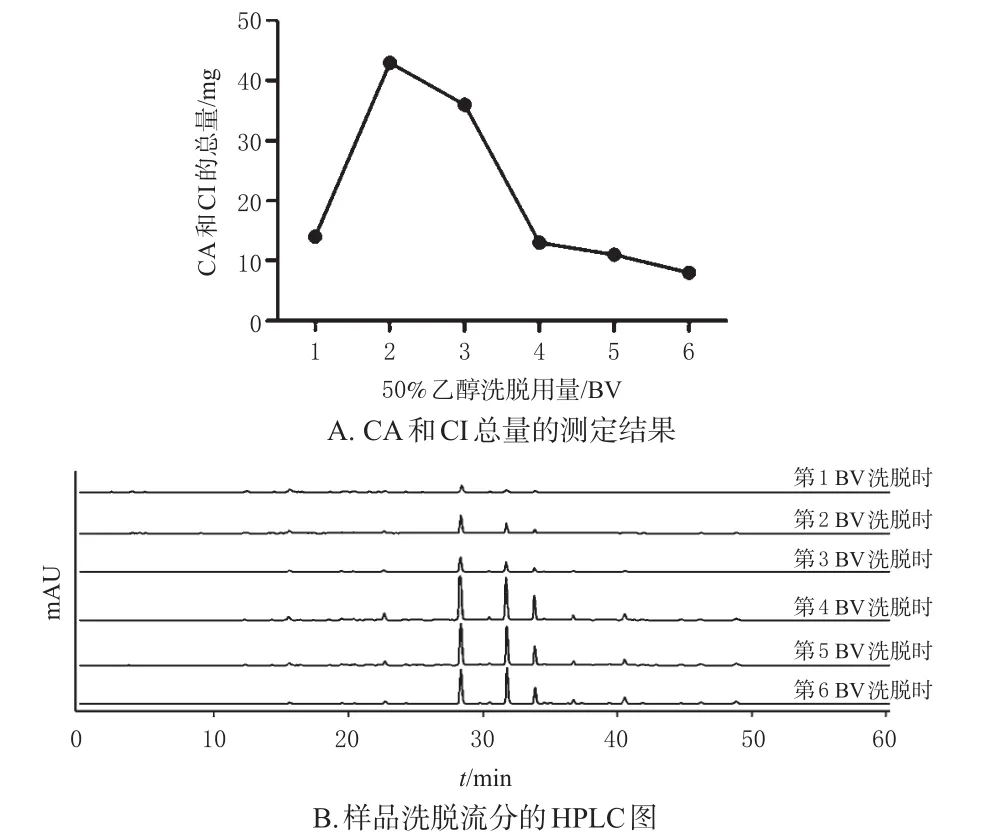
图4 目标产物洗脱剂用量对CA和CI洗脱效果的影响
2.3.6 最优富集工艺的验证 取已标定CA和CI总含量为27.8 mg/g的样品适量,按“2.3.2”项下方法制备上柱样品溶液,分成3份,每份100 mL,分别加入到3份装有D101型大孔吸附树脂的层析柱中,上样流速为2 BV/h,静置1 h;采用上述优化的富集参数进行层析操作,采用HPLC法测定各样品洗脱流分中CA和CI的含量,并计算富集倍数。结果显示,经D101型大孔吸附树脂富集后的提取物中CA和CI的平均总含量为322.7 mg/g,RSD为1.05%(n=3),平均富集倍数为11.61倍,这表明该最优富集工艺的富集效果显著,工艺重复性好。
3 讨论
黄酮类化合物在植物体内分布极其广泛,是许多中草药的有效成分。在植物黄酮类成分的提取方法中,传统的回流提取法设备简单,但提取温度高且提取时间较长,容易造成黄酮类成分的氧化损失;超声提取法与回流提取法相比,能极大缩短提取时间,减少溶剂使用和降低能耗[5―6]。笔者在前期研究中,考察了超声提取法和传统回流提取法对长梗金腰中CA和CI的提取效果,结果发现,超声提取法的提取率较高,且提取时间明显短于回流提取法。笔者进一步对超声提取法的工艺参数乙醇体积分数(0、25%、50%、75%、95%)、料液比(1∶5、1∶10、1∶20、1∶30,g/mL)、提取温度(30、40、50、60、70 ℃)、提取时间(30、45、60 min)进行了单因素实验。结果发现,超声提取时间在45 min以上对CA和CI的提取率没有明显影响。因此,笔者在单因素实验的基础上,进行了4因素3水平的正交实验,以进一步优化长梗金腰中CA和CI的超声提取工艺。结果显示,长梗金腰中CA和CI的最优提取工艺为8倍量的50%乙醇在室温下一次性超声提取45 min。经验证,在该最优提取工艺条件下,CA和CI的转移率可达到95.43%。
大孔吸附树脂是一类人工合成的具有大孔结构的有机高分子共聚体,因其物理化学稳定性高、吸附容量大、选择性好、使用周期长等优点而被广泛应用于天然产物的富集分离[7―9]。采用大孔吸附树脂柱层析工艺富集中药有效成分的关键环节包括树脂类型的筛选、饱和吸附量的确定、杂质洗脱剂考察及其洗脱体积的确定、目标成分洗脱剂考察及洗脱体积确定。前期研究中,笔者通过静态吸附和动态洗脱实验,优选了D101型大孔吸附树脂作为富集长梗金腰中CA和CI的树脂类型。在此基础上,本研究优化得CA和CI的最优富集工艺:将上柱溶液加入D101型大孔吸附树脂柱,静置1 h;用4 BV 20%乙醇洗脱杂质,再用4 BV 50%乙醇洗脱CA和CI。进一步验证,该富集工艺所得CA和CI的总含量为322.7 mg/g,树脂富集倍数为11.61倍。
综上所述,本研究成功优化了长梗金腰中CA和CI的提取富集工艺,可为CA和CI的药物开发利用提供参考。
