电解液调控策略提升水系锌离子电池正极材料电化学性能
2023-03-15齐亚娥夏永姚
齐亚娥 ,夏永姚
1复旦大学化学系,上海市分子催化和功能材料重点实验室,上海 200433
2河西学院化学化工学院,甘肃省河西走廊特色资源利用重点实验室,甘肃 张掖 734000
1 引言
可充电电池作为清洁能源,是新能源领域的一项关键技术,可为大规模电子仪器、设备和交通工具提供能量储存和转换,对实现碳中和起到积极作用。锂离子电池(LIBs)因具有能量密度高、循环寿命长等优点,广泛应用于各种电子设备,但锂储量有限、价格昂贵,其使用的有机电解液导电性低,且存在易燃易爆的安全性问题1,2。因此,研究者们开始寻求能量储存的替代品。
水系电池以水溶液为电解液,与有机电解液相比,表现出安全性高、成本低、环境友好及封装简单等显著的优点,且水溶液的离子电导率高使其具有优异的倍率性能1。近年来,水系二次电池包括单价离子(Li+,Na+,K+,NH4+)3-6和多价离子(Ca2+,Mg2+,Al3+,Zn2+)7-10电池,引起研究者们的广泛关注。其中,水系锌离子电池(ZIBs)因负极锌具有成本低、电极电位低(-0.76 Vvs.SHE)、质量比容量高(820 mAh·g-1),及其在水溶液中良好的稳定性等特点而发展迅速11,12,在大规模电网储能以及柔性和可穿戴电子设备等方面具有广阔的应用前景13,14。
水系锌离子电池通常可分三类:(1)传统的碱性锌镍电池Zn-Ni(OH)2,始于19世纪,具有很长的发展历史,其显著优点是具有高的工作电压(~1.65 V)和能量密度,但差的循环性能是限制其应用的关键因素15。(2)二次混合水系电池(ReHAB)是以常用的锂离子正极材料LiMn2O4或LiFePO4为正极,金属锌为负极,使用锂盐/锌盐混合电解液,充放电过程中正极发生Li+的嵌入/脱出反应,负极为Zn2+的溶解/沉积,电解液体系中Zn2+并未参与正极材料的嵌入反应16,17。(3)“摇椅”型水系ZIBs,正极为Zn2+(或Zn2+/H+,Zn2+/M+)的嵌入/脱出,负极发生Zn2+溶解/沉积。Shoji等18最先分别以MnO2和金属锌为正负极,采用中性或弱酸性水系电解液,提出可充电“锌离子电池”,康飞宇老师课题组之后研究表明MnO2大隧道结构可以储存Zn2+,表现出较高的容量和反应动力学19。然而,在碱性电解液中,负极锌存在严重的腐蚀、枝晶、形变、析氢及因生成ZnO而表面钝化,降低电池的库伦效率和循环寿命。相对于碱性体系,中性和弱酸性电解液更适合于水系ZIBs,锌负极的可逆性得到很大提升,但同样面临电化学稳定窗口较窄、锌枝晶、析氢和副反应等问题。
近年来,随着水系ZIBs的迅速发展,大量性能优异的正极材料不断涌现,包括不同晶型的MnO220-22,普鲁士蓝及类似物23,24,钒基化合物25-27和有机化合物28。但这些正极材料都存在不同程度的问题,其性能有待进一步提升:(1)过渡金属氧化物正极材料在水溶液中的溶解,例如锰基氧化物和钒基化合物在中性或弱酸性水溶液中的溶解29,30;(2)正极材料/电解液界面副反应。部分水系ZIBs正极材料的储存机制属于Zn2+/H+共嵌,在H+嵌入/脱出过程中伴随碱式锌盐(Zn4SO4(H2O)6·4H2O、Znx(CF3SO3)y(OH)2x-y·nH2O和Zn4ClO4(OH)7等)的生成与分解22,31,32,还存在不可逆Zn2(V2O7)(OH)·nH2O副产物的生成33,34;(3)嵌入Zn2+和主体材料之间强的静电作用力造成结构坍塌。这些将会导致循环过程中容量的严重损失,甚至失效,从而影响电池的循环寿命。一方面,正极材料结构设计包括层状化合物的预插层、缺陷工程、表面包覆及修饰22,34-36,对提升ZIBs电化学性能起到了非常积极的作用。另一方面,电解液优化调控也是解决上述问题的有效策略,对改善正极材料电化学性能起到了关键作用37-40。
因此,本文关注电解液调控策略对水系ZIBs正极材料电化学性能的提升,将电解液设计分为溶质(包括锌盐、添加剂和高浓盐)和溶剂调控两大方面,系统总结了电解液优化调控策略对抑制正极材料溶解、提高电位、抑制正极材料/电解液界面副反应等方面起到的积极作用,分析了该策略的提升机制及存在的问题,并展望了其发展方向。
2 电解液基本性质
水分子作为水系电解液的核心,不仅保证了水系ZIBs的高安全性和可逆性,同时决定体系的电化学稳定窗口(electrochemical stability window,ESW)。如图1a所示,纯水发生析氢(HER)和析氧反应(OER)对应的热力学分解电压即ESW仅为1.23 V,而且水分解具有一定的pH依赖性,图1b为HER和OER与电解液pH值之间的关系,在水系ZIBs中,由于涉及多电子转移过程,反应动力学缓慢引起的过电位使水分解的实际电压约为2 V。然而,水系电解液中仍不可避免地发生OER (方程1)和HER (方程2),会极大的降低电池的库伦效率,并限制高电压正极材料的使用。因此,通过增加电解液浓度、增加电解液中阴阳离子或溶剂分子与自由水之间的相互作用力,以降低自由水活性,可拓宽电化学稳定窗口41,42。

图1 (a)电解液中水的析氢析氧反应和ESW 41;(b)Zn-H2O体系在25 °C下的Pourbaix图;(c)Zn2+溶剂化结构及自由水中的氢键Fig. 1 (a)Hydrogen and oxygen evolution reaction of water in aqueous electrolyte; (adapted from Ref. 41).(b)Pourbaix diagram of Zn-H2O system at 25 °C. (c)Solvation structure of Zn2+ and H-bonds of free water.
此外,Zn2+会和周围的水分子之间产生强的相互作用力,在水溶液中以水合离子[Zn(H2O)6]2+的形式存在,其半径(0.860 nm)远大于裸Zn2+(0.148 nm),而体系中自由水分子间则通过氢键(O…H—O)结合成网络结构43(图1c)。因此,电解液调控主要是通过调节溶质和溶剂,改变Zn2+-溶剂化结构,降低Zn2+-溶剂化结构中水分子的数量,破坏水分子间氢键以降低自由水活性。
3 溶质组分调控
3.1 低浓度电解液
3.1.1 锌盐中不同阴离子对正极材料电化学性能的影响
在水系锌离子电解液体系中,除水分子间氢键、Zn2+-H2O之间的作用力外,还存在正-负离子、负离子-H2O之间的作用力。因离子带有电荷,阴离子和水之间的作用力比水分子间氢键强,可以破坏水分子之间氢键网络结构,参与Zn2+的溶剂化过程44。因此,不同阴离子会极大地影响电池的电化学性能,常用的锌盐有ZnSO4、ZnCl2、Zn(NO3)2、Zn(ClO4)2、Zn(CH3COO)2、Zn(CF3SO3)2和Zn(TFSI)2。
Wang等45通过比较ZnSO4、ZnCl2、Zn(NO3)2、Zn(CH3COO)2和Zn(CF3SO3)2等5种相同浓度锌离子电解质混合电容器的电化学交流阻抗(EIS)发现,Zn2+扩散阻抗大小顺序为:ZnCl2<Zn(CF3SO3)2< ZnSO4< Zn(CH3COO)2< Zn(NO3)2,表明ZnCl2电解液中Zn2+在电极材料/电解液界面迁移速率最快,同时ZnCl2电解液也具有最高的离子电导率。然而,1 mol·L-1ZnCl2电解液的ESW窄,充电电位较低(~0.75 Vvs.Zn2+/Zn),导致在较高电位充电时电解液会发生持续的分解(图2a),限制了其在低浓度时的应用46。此外,值得注意的是,与相比,的电位曲线明显不同,主要是因为阴离子具有强氧化性,可以腐蚀锌负极并造成局部甚至整体电解液pH的增加,使CuHCF的容量急剧下降(图2b,c)44,47。其中,ZnSO4和Zn(CF3SO3)2是最常用的两种锌盐,对应的ESW分别能达到2.3和2.4 V (图2d)46,ZnSO4因结构稳定、且具有明显的价格优势,而广泛使用,但是它也不可避免的存在析氢反应,并随着Zn2+/H+共嵌,会在正极材料/电解液界面生成副产物(Zn4SO4(OH)6·4H2O)22,48。与之相比,Zn(CF3SO3)2中阴离子体积庞大,能减少Zn2+周围自由水的数量,减小溶剂化作用,且Zn2+在Zn(CF3SO3)2电解液中具有更高的可逆性和更快的反应动力学,正极材料在Zn(CF3SO3)2中具有较好的电化学性能,其容量、循环稳定性及低温性能均优于ZnSO4体系(图2e,f)49-51。此外,锌盐中阴离子对水中氢键具有重要的影响,图2g比较了几种阴离子的静电势(electrostatic potential, ESP),其大小顺序为,表明和H2O之间的作用力最弱,因此,使用2 mol·L-1Zn(CF3SO3)2为电解液组装的Zn//V2O5电池在-30 °C时仍具有优异的循环性能和容量,在0.5 A·g-1的电流密度下循环1000圈容量保持率为81.7%51。另外,高离液序列的对水分子间氢键具有强的破坏作用52,53,在-60 °C时仍然具有较高的离子电导 率 (1.3 × 10-3S·cm-1), 因此,以 3 mol·kg-1Zn(ClO4)2为电解液的ZIBs具有优异的低温性能,在-60 °C时基于锌离子的混合电容器能呈现室温74.2%的容量,在-30 °C、1 A·g-1电流密度下具有70000圈(280天)的超长循环寿命53。表1列出了常用锌盐的基本特性38,45,47,49,52-55。

图2 (a)Zn电极在1 mol·L-1 ZnCl2电解液中的CV曲线46;CuHCF (实线)和锌负极(虚线)在不同电解液中的电位曲线:(b) 20 mmol·L-1 Zn(ClO4)2和(c) 20 mmol·L-1 Zn(NO3)2 47;(d) Zn电极在1 mol·L-1 ZnSO4和Zn(CF3SO3)2电解液中的CV曲线46;(e) V3O7·H2O正极材料在不同电解液中的循环寿命50;(f) Zn//V2O5电池在两种电解液中的倍率性能比较;(g)不同阴离子的ESP 51Fig. 2 (a) CV curve of Zn electrode in1 mol·L-1 ZnCl2 electrolyte 46. The potential profiles of CuHCF (solid line) and Zn foil (dashed line) at 1C in (b) 20 mmol·L-1 Zn(ClO4)2 and (c) 20 mmol·L-1 Zn(NO3)2 47. (d) CV curves of Zn electrodes in the different electrolytes (1 mol·L-1 ZnSO4 and Zn(CF3SO3)2) 46. (e) Cycle life of the V3O7·H2O cathodes at 0.5 A·g-1 with different electrolytes 50. (f) Rate capability of Zn//V2O5 in the two electrolytes. (g) ESP mapping of the different anions51.

表1 不同锌盐水系电解液的基本性质Table 1 The characteristic of different zinc salt in aqueous electrolyte.
3.1.2 添加剂对正极材料性能的提升
3.1.2.1 溶解-平衡抑制正极材料溶解
锰基与钒基氧化物是水系ZIBs常用的正极材料,在中性及弱酸性电解液中存在不可避免的溶解问题,循环过程中容量急剧衰减30,48,56,57。在电解液中预添加适当的金属阳离子,根据正极材料和电解液中活性物质的化学溶解-平衡原理可以有效抑制正极材料的溶解,从而延长电池的循环寿命25,48,58-62。如图3a所示,在ZnSO4电解液中MnO2正极容量严重衰减,尤其是前10圈,对电解液中Mn2+浓度检测发现,其浓度增加与电池容量衰减呈对应关系(图3b),说明容量衰减是锰的溶解所致,随着循环的进行,电解液中溶解的Mn和电极材料中Mn达到平衡,衰减速率减慢,因此,向ZnSO4电解液中预添加MnSO4,可通过同离子效应在电解液和正极材料中建立Mn的溶解平衡,以抑制MnO2的溶解(图3c)58。此外,随着添加剂MnSO4用量的增加,电池的容量保持率显著提高,说明添加剂的用量对电极材料性能的提升也起到非常重要的作用(图3d,e)61,62。同时,改变不同的锰盐,也能起到相同的作用,Zhang等59在2 mol·L-1Zn(CF3SO3)2电解液中添加 0.2 mol·L-1Mn(CF3SO3)2,预添加的Mn2+在正极表面生成均匀的多孔MnOx纳米片进而抑制Mn的溶解。
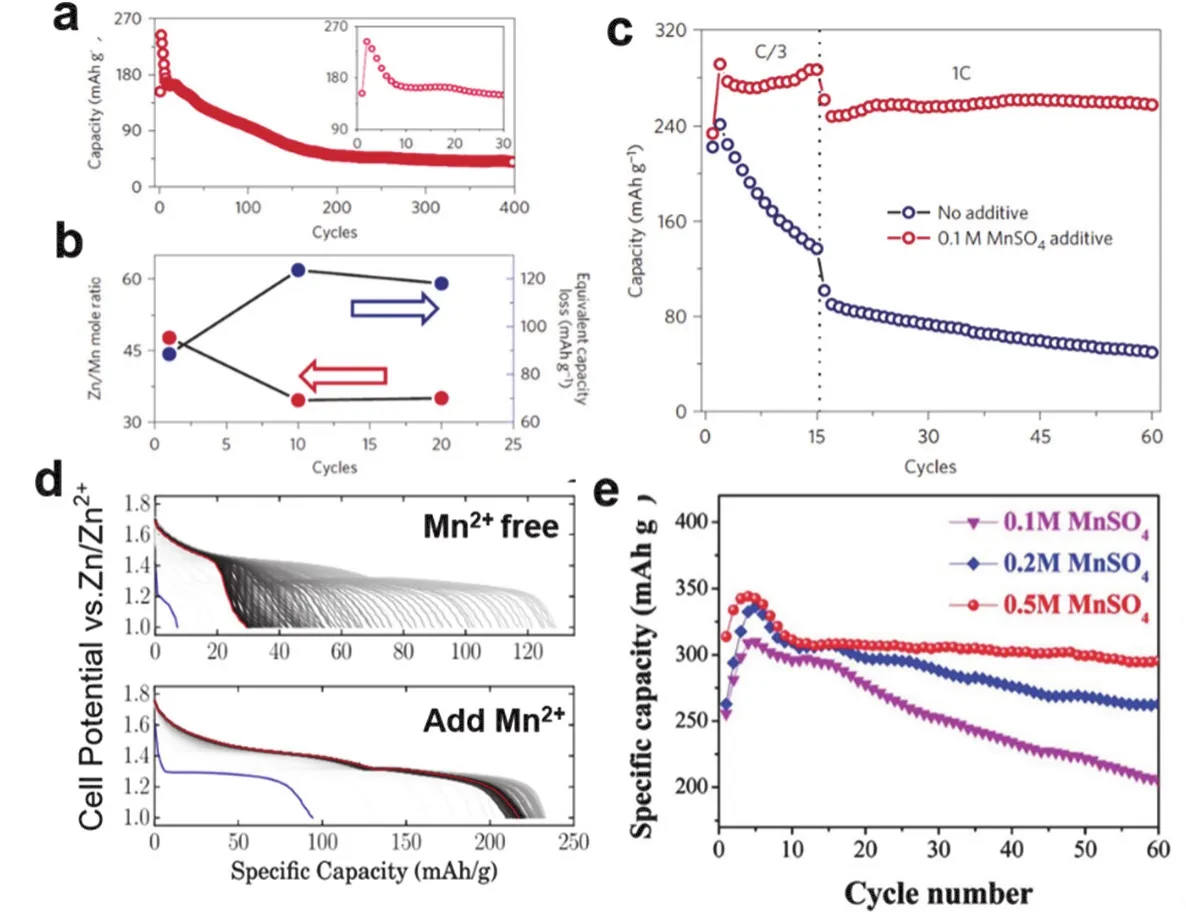
图3 Zn//MnO2电池的循环性能Fig. 3 The cycle performance of Zn//MnO2 battery.
对于钒基氧化物,金属离子插层钒氧化物在嵌锌过程中,插层离子可从层状结构中脱出,呈现置换/插层机理63-65。电解液中添加预插层金属离子根据同离子效应可在电解液与材料间形成溶解-平衡,阻止插层离子从层间脱出,维持结构稳定性48,66,67。从图4a可以看出,NaV3O8·1.5H2O在1 mol·L-1ZnSO4电解液中容量迅速衰减,结合内插图,可发现主要原因是正极材料在1 mol·L-1ZnSO4中的溶解所致,向其中添加Na2SO4之后,材料的溶解性及电池的循环性能明显改善(图4a,b),在4 A·g-1电流密度下循环1000圈能有82%的容量保持率48。此外,在ZnSO4电解液中添加MgSO4,则能阻止MgxV2O5·nH2O正极材料在循环过程中发生层间Mg2+的脱出,同时可抑制钒的溶解,以稳定结构,提升容量及循环寿命,同样,添加剂的量不同,其容量也稍有差别,而且Mg2+也能嵌入到正极材料层间,属于Zn2+/Mg2+共嵌机制,在充电过程中,电解液中的Mg2+与Zn2+一同沉积于负极侧,添加Mg2+对容量有一定的贡献(图4c,d)67。此外,通过两种电解液中循环后Zn负极的电镜照片可以看出,添加Na2SO4后Zn负极表面光滑平整,能有效抑制锌枝晶的生长(图4e),说明Na2SO4添加剂能同时抑制正极材料的溶解和负极锌枝晶的生长(图4f)48。
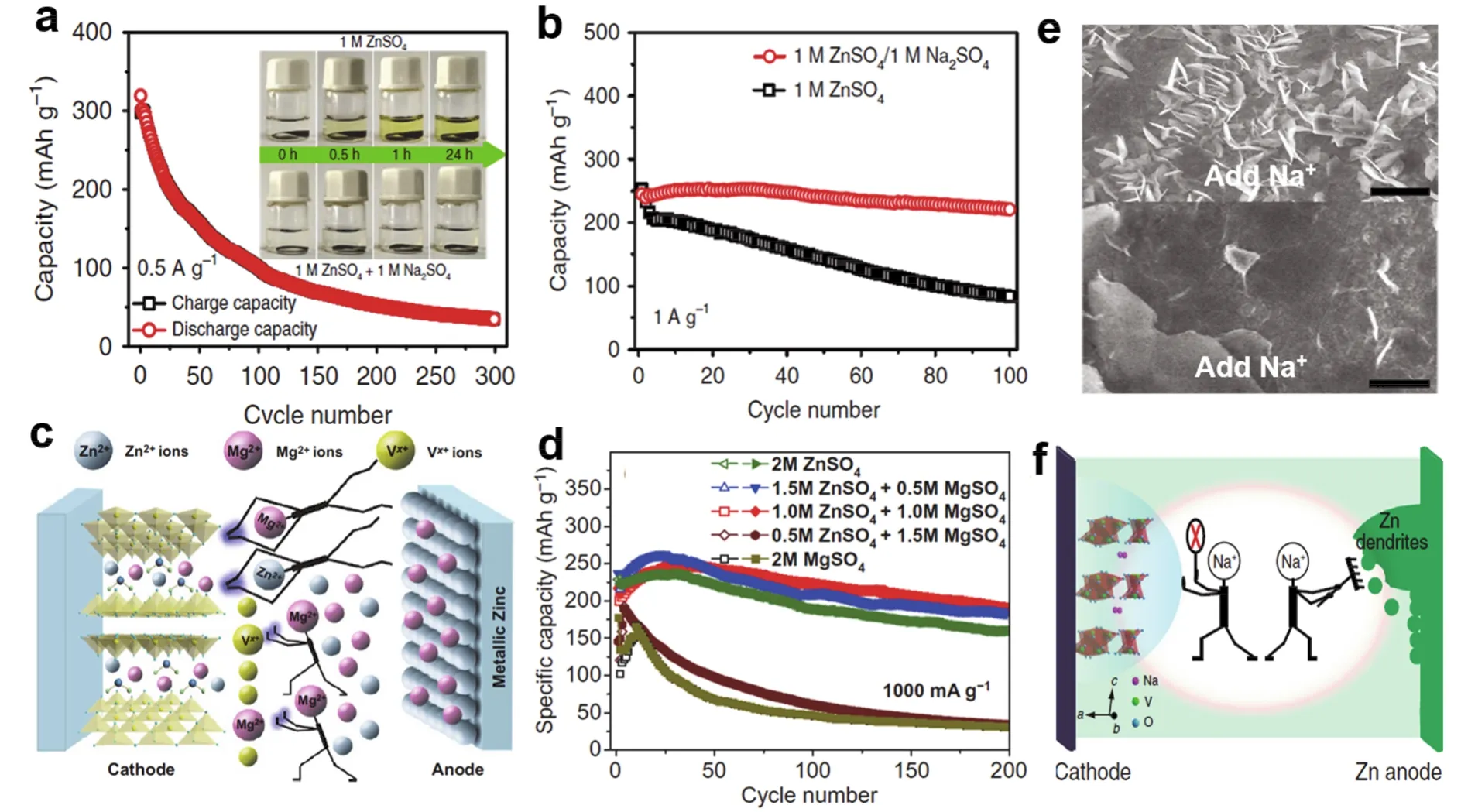
图4 (a)NaV3O8·1.5H2O(NVO)电极在1 mol·L-1 ZnSO4中的循环性能(内插图为NVO极片在两种电解液中浸泡不同时间的光学照片),(b)NVO电极的循环性能48;(c)Mg2+添加剂在电解液中的作用示意图,(d)不同电解液中Zn//MgxV2O5·nH2O的循环性能比较67;(e)电解液中添加Na2SO4前后Zn负极循环后的SEM照片,(f)添加Na2SO4对抑制正极材料溶解和锌枝晶的作用示意图48Fig. 4 (a)Cycle performance of NaV3O8·1.5H2O (NVO)electrode in 1 mol·L-1 ZnSO4 (The insets are optical photographs of NVO electrodes in the two electrolytes for different periods). (b)Cycle performance of NVO electrode in the two electrolytes 48. (c)Schematic of Mg2+ addictive functional mechanism and (d)cycle performance of Zn//MgxV2O5·nH2O battery with different electrolytes 67. (e)SEM images of Zn anodes after cycle with or without Na2SO4. (f)Schematic illustration of Na2SO4 additive for inhibiting dissolution of cathode and growth of Zn dendrites 48.
3.1.2.2 提高电池的工作电压
在电解液中添加适当的无机酸、盐或表面活性剂,可以提高电池的工作电压68-73。Chao等68通过简单方法设计的Zn//MnO2电池,在ZnSO4+MnSO4混合电解液,电池放电分三个阶段(图5a),分别表示为D1、D2和D3,其中D1是2电子反应,D2和D3均为单电子反应,随着充放电的进行,D1放电平台逐渐延长(图5b),且在此过程中pH逐渐降低,前10圈pH由4.60降低至2.32,这可能是容量及电压平台逐渐升高的原因。因此,在混合电解液中添加不同浓度的H2SO4,改变体系pH值,从而使放电平台仅保留D1的2电子反应阶段,可以将Zn//MnO2电池的工作电位提高至1.95 V,其容量也高达507 mAh·g-1,电压和容量均远高于传统Zn//MnO2电池(图5c)。层状VOPO4锌电正极材料因具有较高的工作电压而被广泛关注,但遗憾的是,在3 mol·kg-1Zn(CF3SO3)2水系电解液中,VOPO4会分解为VOx和,使VOPO4·xH2O在循环后电压严重衰减,放电曲线形状类似VOx,向此电解液中添加0.8 mol·kg-1H3PO4,根据同离子效应可抑制VOPO4的分解,的存在维持了VOPO4的原有高电压69。然而,H2SO4或H3PO4的加入,在提高电压的同时,会使电解液体系酸性增大,将不利于负极Zn的长循环。另外,抑制VOPO4·xH2O分解作用的是,因此,用含有的锌盐替代H3PO4,是否也能起到相同的作用并避免H+浓度发生大的变化?近期,孙筱琪课题组70进一步在2 mol·L-1ZnCl2水系电解液中添加0.07 mol·L-1Mn(H2PO4)2,能将电池的工作电压提高至1.75 V,与含有MnCl2的体系相比高出0.4 V (图5e),在放电阶段正极表面生成Zn3(PO4)2·4H2O,释放的质子促进MnO2还原为Mn2+,充电时Zn3(PO4)2·4H2O又质子化为Zn(H2PO4)2,该过程是完全可逆的(图5d)。Zn3(PO4)2·4H2O/Zn(H2PO4)2起到储蓄质子并缓冲局部pH变化的作用,原位pH测试表明在10圈循环过程中电解液pH的变化非常小。结合之前Chao等68的研究结果,该体系电位的提高应该主要是H+起作用,但同时避免了局部pH的剧烈变化,这对电解液多功能化设计提供了新的思路。

图5 (a)Zn//MnO2在1 mol·L-1 ZnSO4/1 mol·L-1 MnSO4混合电解液中的放电机理,(b)混合电解液中不同循环阶段的放电曲线,(c)添加不同量H2SO4后的放电曲线68;(d)Zn//MnO2电池正极工作机理;(e)Zn//MnO2电池在添加0.07 mol·L-1 Mn(H2PO4)2前后的放电曲线70;(f)第二圈的充放电曲线(1 mol·L-1 Zn(CF3SO3)2,1 mol·L-1 Zn(CF3SO3)2/1 mol·L-1 Al(CF3SO3)3)71;(g)Co3O4电极的充放电曲线(1 mol·L-1 KOH, 2 mol·L-1 ZnSO4 + 0.2 mol·L-1 CoSO4)73Fig. 5 (a)Schematic of charge storage mechanism of Zn//MnO2 battery in 1 mol·L-1 ZnSO4/1 mol·L-1 MnSO4 electrolyte. Discharge step at different cycles (b)in 1 mol·L-1 ZnSO4/1 mol·L-1 MnSO4 electrolyte and (c)with various concentration of H2SO4 68. (d)Schematic illustration of storage mechanism of Zn//MnO2. (e)Discharge profiles of Zn//MnO2 cells before and after add Mn(H2PO4)2 70. (f)The second charge/discharge curves of ZIBs(1 mol·L-1 Zn(CF3SO3)2 and 1 mol·L-1 Zn(CF3SO3)2/1 mol·L-1 Al(CF3SO3)3)71. (g)Charge/discharge curves of Co3O4 cathode (1 mol·L-1 KOH, 2 mol·L-1 ZnSO4 + 0.2 mol·L-1 CoSO4)73.
除了上述改变pH值或同离子效应能提高电池的工作电压之外,混合离子(双阳离子)电解液也是提高正极材料工作电压的有效策略。如,Li等71以MnO2为正极组装的水系ZIBs,使用双离子(1 mol·L-1Zn(CF3SO3)2/1 mol·L-1Al(CF3SO3)3)混合电解液,获得1.7 V的高工作电压,比单离子电解液中电位高出约0.3 V (图5f),机理研究表明,与上述不同的是,H+对工作电位几乎没有影响,放电平台的提高是在放电过程中生成了层状结构的AlxMnO2·nH2O所致72。Ma等73报道的富Co(III)-Co3O4纳米棒水系ZIBs,在ZnSO4中添加CoSO4,使电解液pH接近中性,与传统使用的碱性电解液相比,电压窗口可扩宽至2.2 V,工作电压提高至2.1 V (图5g),且该体系具有较高的容量(0.5 A·g-1时205 mAh·g-1)及优异的循环性能(4 A·g-1下循环5000圈容量保持率为92%)。此外,添加表面活性剂十二烷基硫酸钠也可以提高电池的工作电压74。
另外,电池工作电压的高低与添加离子和锌离子之间的相对含量有关,Huang等23研究表明,以ZnHCF为正极组装ZIBs,在ZnSO4中添加K2SO4,随着Zn2+/K+比值的减小,CV曲线中高电位还原峰正移,且强度增强,低电位还原峰的强度则逐渐减弱,说明还原峰正移(工作电位的提高)是K+在电极材料中的选择性嵌入所致。但是,工作电位最高的电解液体系 (0.05 mol·L-1ZnSO4+ 0.5 mol·L-1K2SO4),因Zn2+浓度太低ZnCHF正极材料易溶解,导致容量衰减最快,因此,将两者浓度调节至合适比例,则能同时兼顾工作电压和容量保持率。
3.1.2.3 正极材料/电解质界面膜(CEI)保护正极材料
通过添加电解质盐或有机小分子,可以在正极材料/电解液界面原位生成界面保护膜,抑制电极材料的进一步溶解,达到保护正极材料的目的。Xu等75以Na0.55Mn2O4·nH2O为正极材料,在混合电解液2 mol·L-1ZnSO4+ 0.1 mol·L-1MnSO4中进一步添加NaSO4,研究发现Na+的添加不仅可以通过修饰锌离子的沉积模型抑制锌枝晶的生长,同时从第2、3圈充电阶段开始,可以在正极材料表面生成不可逆纳米花状产物Na12Mn7(SO4)13·15H2O(JCPDS No. 29-1248),可以阻止锰溶解并维持结构稳定性。同样,在ZnSO4中加入适当的强场配体尿素,则可生成新的溶剂化结构[Zn(H2O)2(urea)3]2+,减少了Zn2+溶剂化结构中水分子的数量,降低了对正极材料结构的破坏,且在正极/电解液界面原位形成ZnCO3保护膜,稳定正极材料并抑制Mn的溶解76。此外,电解液中添加少量70,77,则能生成Zn3(PO4)2·4H2O保护膜,如1 mol·L-1Zn(CF3SO3)2+0.025 mol·L-1Zn(H2PO4)2体系,可以在电极材料表面生成保护膜抑制V2O5正极材料溶解,在0.8 A·g-1的电流密度下循环1000圈,容量保持率仍高达88.1%77。通过以上研究发现,电解液添加剂能有效改善正极材料电化学性能:提高工作电压、容量,延长循环寿命。但是,不同添加剂对电解液溶剂化结构和电化学行为的影响机制尚不是很清楚,Sun等28通过密度泛函理论(DFT)计算了(0.21 nm)和(0.22 nm)溶剂化结构中金属离子和水的结合能,结果表明Mg2+和水的结合能更小,通过Mg2+和水结合以破坏水分子间氢键,但未见Mg2+参与到Zn2+的溶剂化结构中,因此添加剂对Zn2+溶剂化结构有没有影响、以及如何影响?其机制还有待进一步研究。而且正极材料和电解液之间的界面兼容性和普适性并不是很高,需进一步开发更多的离子添加剂,深入研究其与电极材料之间的作用机制。
3.2 高浓度电解液(Water-in-salt electrolyte,WiSE)
[Zn(H2O)6]2+溶剂化结构中,Zn2+和水之间的结合作用强,脱溶剂化需克服很高的能垒76。提高电解液浓度,一方面可以减少Zn2+溶剂化结构中的水分子,重建Zn2+溶剂化结构78-80。如图6a所示,在稀溶液LiTFSI中,Li+的原始溶剂化结构中周围结合4个水分子,当电解液中LiTFSI含量增高时,溶剂化结构中水分子逐渐降低至2.5个79。同样,将Zn(TFSI)2与高浓度LiTFSI混合后,可以改变Zn2+的溶剂化结构,如图6b所示,当Zn2+处于稀溶液状态(1 mol·kg-1Zn(TFSI)2+ 5 mol·kg-1LiTFSI)时,Zn2+结合6个水分子[Zn(H2O)6]2+,中等浓度(1 mol·kg-1Zn(TFSI)2+10 mol·kg-1LiTFSI),阴离子TFSI-开始进入Zn2+的溶剂化鞘结构,当达到高浓度(1 mol·kg-1Zn(TFSI)2+ 20 mol·kg-1LiTFSI)时,Zn2+溶剂化鞘结构几乎全部被TFSI-占据80。另一方面,高浓度电解液中,由于阴阳离子之间、离子-水分子之间等作用力的存在,能破坏水分子之间的氢键网络结构,从而降低自由水的活度(图6c)81。Zn2+溶剂化结构和自由水活度这两个因素是电解液调控优化、提升电池电化学性能的关键。因此,可以使用溶解度较大的锌盐(ZnCl2、ZnBr2)81-83,或将溶解度较大的其他金属盐引入水系锌离子电解液体系,包括LiTFSI、LiCF3SO3、NaClO4和CH3COOK等80,84-86。
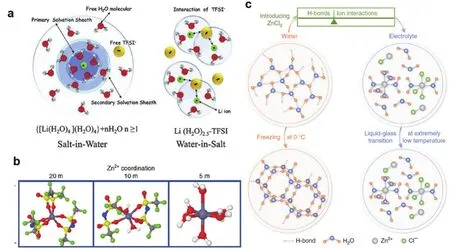
图6 (a)稀溶液和WiSE中的Li+溶剂化结构的演变过程79,(b)三种浓度下Zn2+的溶剂化结构80;(c)水溶液和WiSE状态下Zn2+的溶剂化结构及水分子间氢键和低温电解液设计81Fig. 6 (a)Illustration of the development of the Li+ solvation sheath in diluted and WiSE 79.(b)The representative Zn2+ solvation structures in the three concentrations of 1 mol·kg-1 Zn(TFSI)2 with LiTFSI (5 mol·kg-1, 10 mol·kg-1 and 20 mol·kg-1)80. (c)Schematic of the structure evolution of water and electrolyte, and the design of low-temperature solution 81.
3.2.1 扩宽电化学窗口提高正极材料工作电位
2015年Suo等79最早报道WiSE可以拓宽水系电解液的ESW,有效抑制析氢和析氧反应。自此,一系列WiSE应用于水系电解液,以实现较高的电压和能量密度86-90。根据如下能斯特方程,阳离子活度增加会提高正极材料的工作电位79,91,92:
方程中E°、R、T、n、F分别代表标准电势、气体摩尔常数(8.3145 J·K-1·mol-1)、常温(298 K)、电子转移数(n= 2)和法拉第常数F(96485 J·V-1·mol-1),acation、ccation和γcation分别为阳离子的活度、浓度及活度系数,图7a为不同浓度电解液对体系ESW的影响93。Zhao等94组装的水系混合离子电池Zn/LiMn0.8Fe0.2PO4,采用21 mol·kg-1LiTFSI + 0.5 mol·kg-1ZnSO4为电解液,有效扩宽了电化学窗口,获得183 Wh·kg-1和1.8 V的高能量密度和工作电压。Hu等90使用相似的混合电解液21 mol·kg-1LiTFSI + 1 mol·kg-1Zn(CF3SO3)2,组装的Zn//V2O5电池,放电平台可从0.6 V提升至1.0 V (图7b),同时提高了能量密度和循环性能。该电解液体系也能将VOPO4正极材料电化学窗口拓宽至2.1 V,平均工作电压提升至1.56 V附近,在此工作电压范围,氧的氧化还原变的可逆,且能贡献27%的容量(图7c,d)37,95。除了采用高溶解度的锂盐之外,ZnCl2在水中同样具有很高的溶解度,虽然此电解液在低浓度使用时对电极材料和电池壳并不友好,但当浓度增大则可发挥良好的性能。普鲁士蓝类正极材料(Zn3[Fe(CN)6]2)在30 mol·kg-1ZnCl2电解液中,Zn2+的嵌入电位同样得到提升,可高达1.0 V (vs.Ag/AgCl)93,而在稀溶液ZnSO4中其电位约为 0.2 V96; 30 mol·kg-1ZnCl2也可以将Ca0.2V2O5·0.80nH2O正极材料的工作电位提升0.15 V97。

图7 (a)(e)Zn3[Fe(CN)6]2正极和Fc/C负极在不同浓度ZnCl2电解液中的CV曲线93;(b)0.1 A·g-1下Zn//V2O5电池在两种电解液中第15圈的充放电曲线90;(c)Zn负极在两种电解液中的CV曲线;(d)Zn//VOPO4电池在两种电解液中的首圈充放电曲线95Fig. 7 (a)Comparison of CV curves of the Fc/C anode and Zn3[Fe(CN)6]2 cathode in ZnCl2 electrolytes with different concentration 93. (b)The 15th cycle charge/discharge profiles of Zn//V2O5 cells at 0.1 A·g-1 with different electrolytes 90.(c)CV curves of Zn electrodes and (d)first charge/discharge profiles of Zn//VOPO4 batteries in the two electrolytes 95.
3.2.2 抑制正极材料溶解
高浓度电解液可以降低自由水的活性,能有效抑制正极材料的溶解,延长其循环寿命96-100。与超高浓度(21 mol·kg-1LiTFSI、21 mol·kg-1NaClO4和30 mol·kg-1ZnCl2)相比,中等浓度的电解液在抑制正极材料溶解方面也起到了积极作用。Bin等98使用高浓度电解液(15 mol·kg-1NaClO4+ 1 mol·kg-1Zn(CF3SO3)2)有效抑制了Zn//C-NaVPO4F电池中钒的溶解,使其在1 A·g-1电流密度下循环4000圈,仍能保持89.3%的容量。如前所述,VOPO4·xH2O正极材料在 3 mol·kg-1Zn(CF3SO3)2)中加入 0.8 mol·kg-1H3PO4,虽有效抑制了VOPO4的分解,但并不能抑制其在电解液中的溶解,使用13 mol·kg-1ZnCl2+ 0.8 mol·kg-1H3PO4电解液体系同时解决了正极材料分解和溶解的问题,电解液体系中不同组分对材料的作用如图8a所示,根据同离子效应抑制材料的分解,而13 mol·kg-1ZnCl2则起到抑制材料溶解的作用69。此外,与较低浓度(5 mol·kg-1和10 mol·kg-1)相比,中等浓度的15 mol·kg-1ZnCl2能显著抑制K0.486V2O5正极材料的溶解,在3 A·g-1下其循环寿命可长达1400圈(图8b)99。同时,Liu等85通过表征循环后正极材料的形貌及隔膜颜色变化,进一步说明WiSE能有效抑制正极材料溶解,Zn//VO2电池在2 mol·L-1Zn(CF3SO3)2中循环后,因钒的溶解,隔膜颜色发生变化,且随着循环的进行,颜色逐渐变深,而在2 mol·L-1Zn(CF3SO3)2+ 8 mol·L-1LiCF3SO3体系中,隔膜颜色并未发生明显变化,结合SEM表征发现,在低浓度电解液中循环100圈(0.2 A·g-1)后,VO2形貌变成不均匀的碎片状,高浓度电解液中则能基本保持材料形貌原状。
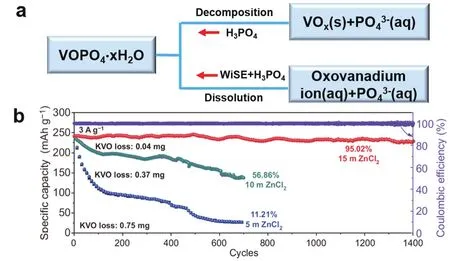
图8 (a)VOPO4分解或溶解路径示意图;(b)K0.486V2O5电极的循环稳定性(3 A·g-1)99Fig. 8 (a)Proposed VOPO4 degradation pathways in different aqueous electrolytes (adapted from Ref. 69).(b)Cycle stability of K0.486V2O5 cathode in three electrolytes at 3 A·g-1 99.
3.2.3 改变储存机制抑制界面副反应
水系ZIBs正极材料的储存机理包括嵌入/脱出机制、转化机制、置换/嵌入机制、溶解-沉积机制等。其中Zn2+/H+共嵌机制是材料获得高容量的重要原因,但是,伴随H+的嵌入,正极材料表面会生成大量的可逆碱式锌盐或不可逆副产物,将增大界面阻抗,阻止Zn2+在正极材料中的进一步嵌入,影响其容量和寿命;另一方面,大尺寸[Zn(H2O)6]2+的嵌入,会造成材料结构急剧膨胀甚至坍塌。高浓度电解液可以裁剪或改变储存机制,使共嵌机制变为单一离子嵌入为主导或脱溶剂化之后再嵌入101,102,能防止结构因过度膨胀而坍塌,且可以改变正极材料/电解液界面接触状态,有效抑制副反应97,103。
V2O5在稀溶液 (1 mol·L-1ZnSO4)中 为H2O/Zn2+共嵌,[Zn(H2O)6]2+半径大,嵌入过程中V2O5经历严重的晶格膨胀,层间距从0.44 nm膨胀至1.39 nm,会引起结构塌陷(图9a);高浓盐(30 mol·kg-1ZnCl2)中,Zn2+在V2O5中的扩散能垒远高于H+,限制了Zn2+在V2O5中的嵌入,使H+嵌入占据主导地位,嵌入质子尺寸小,避免了V2O5晶格的严重扩张,维持了结构稳定性,因此,其在WiSE中循环300圈之后容量仅损失了60.6 mAh·g-1(图9b)101。此外,在低浓度ZnSO4电解液中,质子嵌入的同时会在正极材料表面生成副产物Zn4SO4(OH)6·3H2O22,48,75,随着循环的进行副产物逐渐变得不可逆,这是MoO3正极材料在ZnSO4中循环性能差的主要原因之一102。在30 mol·kg-1ZnCl2电解液中,MoO3正极材料表面生成的放电产物Zn5(OH)8Cl2·3H2O是高度可逆的,且随着循环的进行,放电阶段产物逐渐增多,表示质子嵌入逐渐占据主导地位并高度可逆(同Zn//V2O5),因此,WiSE中MoO3电极具有优异循环性能的两个关键因素是Mo极微小的溶解及质子嵌入的高度可逆性102。Zhang等97以Ca0.2V2O5·0.80nH2O为水系ZIBs正极材料,系统考察了ZnCl2电解液浓度从1 mol·L-1增加至30 mol·kg-1时,电池的容量从296 mAh·g-1提升至496 mAh·g-1,循环稳定性也显著提升。非原位XRD表征发现,在1 mol·L-1ZnCl2电解液中,放电至0.22 V之后,正极表面会生成Zn3(OH)2V2O7·2H2O (图9c),证明钒在稀溶液中的溶解及副产物的生成,将导致活性物质损失,造成容量衰减,而在30 mol·kg-1ZnCl2电解液中,整个放电过程没有观察到造成容量衰减的副产物Zn3(OH)2V2O7·2H2O (图9d)43,97。

图9 (a)V2O5正极在稀溶液和WiSE中的结构演变图示,(b)两种电解液(1 mol·L-1 ZnSO4和30 mol·kg-1 ZnCl2)中不同循环阶段电极的总容量衰减101;Ca0.20V2O5·0.80H2O正极在(c)1 mol·L-1和(d)30 mol·kg-1 ZnCl2电解液中首圈充放电及第2圈放电阶段的非原位XRD谱图97Fig. 9 (a)Illustration of the dynamic structural evolution for V2O5 cathode in dilute electrolyte or WiSE.(b)The total irreversible capacity loss during the cycle at 50 mA·g-1 in different electrolyte (1 mol·L-1 ZnSO4 or 30 mol·kg-1 ZnCl2)101. Ex-situ XRD patterns of the Ca0.20V2O5·0.80H2O cathode in (c)1 mol·L-1 and(d)30 mol·kg-1 ZnCl2 electrolyte of the first discharge/charge and 2nd discharge profiles 97.
除了高浓度电解质之外,有机糖类分子含有大量的羟基,在水中溶解度非常高,且羟基可以破坏水分子间的氢键网络结构并参与阳离子的溶剂化结构103-105。鉴于此,梁叔全课题组103将大量麦芽糖引入到常规电解液中,研究发现,低浓度时水分子除了和Zn2+形成溶剂化结构之外,水分子之间通过大量氢键结合成网络结构,加入大量麦芽糖后,氢键网络被破坏,并参与到Zn2+溶剂化结构的重构,形成更大尺寸(2.665 nm)的配位结构[ZnM3]2+,将水分子有效阻挡在溶剂化壳之外,并通过氢键将自由水和羟基结合,极大降低了自由水的活性。由于[ZnM3]2+尺寸过大,在Zn2+离子嵌入时,先脱溶剂化后嵌入,能保证正极材料的结构完整、并抑制副产物生成及钒的溶解,使Zn//NH4V4O10电池具有长循环寿命,5 A·g-1下可稳定循环4000圈,容量几乎无衰减。
正如凡事都具有两面性,WiSE虽然能拓宽电化学稳定窗口、防止正极材料结构坍塌、抑制其溶解与界面副反应,提高电池的工作电压和循环寿命。但是,因电解液浓度过大,也带来了一些显著的负面问题:(1)粘度大,导致Zn2+离子扩散速率减慢,尤其是大电流密度下,极大影响了电池的倍率性能;(2)固-液转变温度高,低温下盐的溶解度降低,易结晶析出,刺穿隔膜造成电池短路;(3)电解液成本高,高浓度电解液常用盐价格高、且用量大,会极大的增加电池的成本。因此,需进一步开发浓度小于5 mol·kg-1的低浓度电解液,兼顾容量、倍率性能和循环寿命。表2所列为溶质调控对ZIBs正极材料电化学性能的提升。

表2 溶质调控对正极材料电化学性能的影响Table 2 The affection of solute regulation on electrochemical performance of cathode materials.
4 溶剂调控
溶剂是电解液组分的另一重要组成部分,溶剂调控主要是将能与水混溶的有机溶剂,通过不同比例混合,它们对水系ZIBs电化学性能的影响取决于相对含量及其与Zn2+离子之间的溶剂化作用。有机溶剂如乙腈(ACN)108、二甲亚砜(DMSO)109,110、乙醚111、醇类(甲醇、乙醇、乙二醇、丙三醇等)105,112-116、丙烯酰胺(AM)113、1,2-二甲氧基乙烷(DME)117、磷酸三乙酯(TEP)38,118,119、聚乙二醇(PEG400)120等,能破坏Zn2+的溶剂化结构,并参与重组,使溶剂鞘结构中的水分子数量减少,此外,这些溶剂含有电负性较高的O和N,可以与电解液中水分子间形成氢键,从而破坏自由水分子间的氢键网络结构。图10所示为水系电解液体系中添加甲醇,对Zn2+溶剂化结构和水中氢键的影响,可以看出,随着甲醇量的增多,它可以逐渐进入Zn2+的溶剂化结构,并与水分子间形成氢键,且形成氢键的数量与甲醇的添加量呈正相关。对于锌负极,添加有机溶剂可以抑制锌枝晶、析氢反应及副反应,拓宽电池工作温度39,112。如图11a所示,V2O5电极在Zn(CF3SO3)2电解液体系中发生溶解,并与水反应生成不可逆副产物Zn3V2O7(OH)2·2H2O,造成容量的严重衰减,在电解液中加入磷酸三乙酯(TEP),构建了新的溶剂化结构Zn(CF3SO3)2-TEP-H2O,成功抑制了自由水活性及V2O5的溶解,且能阻止副反应发生(图11b),此外,在循环过程中,Zn(CF3SO3)2-TEP-H2O电解液体系能在锌负极侧原位生成SEI (poly-ZnP2O6-ZnF2),从而有效抑制锌枝晶、腐蚀、副反应及HER等问题(图11c,d),因此,Zn//V2O5电池在5 A·g-1的电流密度下循环1000圈后仍有250 mAh·g-1的容量剩余。Wang等121使用高电压VPO4F为水系ZIBs正极材料,在水系电解液中表现出Zn2+/H+共嵌入机制,但两者嵌入阶段不同,高电压1.9 V放电平台对应Zn2+的嵌入,低于1.7 V则属于H+嵌入,在嵌H+阶段有界面副产物Znx(CF3SO3)y(OH)2x-y·nH2O生成,为了抑制H+的嵌入,设计了有机/无机混合电解液体系(2 mol·L-1Zn(CF3SO3)2·2H2O-PC)(PC:碳酸丙烯酯),能有效裁剪嵌入机制,使嵌Zn2+占主导,极大延长1.9 V处的放电平台,且因嵌H+被阻止,副产物的生成也被抑制,电池稳定性得到极大提升,0.2C下循环200圈之后容量保持率仍能维持82.5%,同时能获得高达237 Wh·kg-1的能量密度。Li等122使用Li3V2(PO4)3为正极材料,比较了其在 1 mol·kg-1Zn(ClO4)2水溶液,及 1 mol·kg-1Zn(ClO4)2乙腈和水(ACN-H2O)混合电解液中的电化学行为及性能,如图11e所示,Li3V2(PO4)3电极材料在水系电解液中容量急剧衰减,可能是VOPO4·nH2O在水系电解液中的分解及溶解所致69,在ACN-3% H2O混合体系中循环稳定性显著增加,200圈后仍有87 mAh·g-1的剩余容量,当水含量进一步增加(ACN-11% H2O),电池的初始容量可达125 mAh·g-1,200圈后容量可余121 mAh·g-1,此外,从循环200圈后的充放电曲线可以看出,在纯水系电解液中电极的工作电压也急剧下降,而在混合体系中工作电压高且稳定(图11f)。此外,从图11g-h中H11Al2V6O23.2电极充放电3天后的SEM照片可以看出,在纯水体系(H2O-Zn(ClO4)2)中有大量片状碱式锌盐Zn4ClO4(OH)7生成,而在有机/无机混合体系(PC/H2O-Zn(ClO4)2)中并未观察到,进一步说明混合体系可以有效抑制界面副反应32。
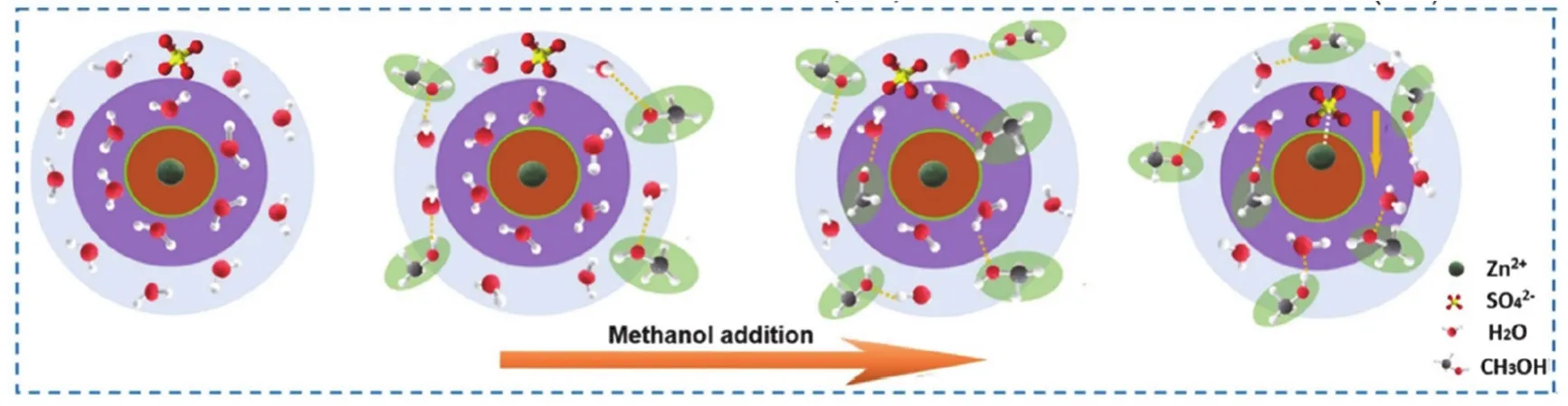
图10 添加不同量甲醇对Zn2+溶剂化结构和水中氢键的影响示意图112Fig. 10 Schematic of development of the Zn2+ solvent sheath and H-bonds with the addition of methanol 112.
进一步对甲醇和乙醇与水混合电解液体系研究发现,有机溶剂在水系电解液体系中的用量对电化学性能有较大影响,虽然这些有机溶剂能与水以任意比例互溶,但锌盐在混合溶剂中的溶解度不同,当甲醇在水中的摩尔比例超过55%时,ZnSO4溶解度降低,会重新结晶析出112,而95%乙醇则能使Na1.87Mn0.78[Fe(CN)6]·2H2O电极5C下的寿命延长至20000圈(时常约1年)116。另外,不同的锌盐在有机/水混合电解液中的溶解性及溶剂化结构也有很大差异123。表3比较了多种有机溶剂的物理常数和添加于水系ZIBs中所起的主要作用,如前所述,溶剂中含有(N、O)原子对体系中Zn2+溶剂化结构、氢键的重组起到非常重要的作用,因此,在选择有机溶剂或添加剂时,溶剂中必须含有高电负性原子(N、O、F),另外,根据所设计电池的性能需求,需合理考虑以下相关联因素:高的粘度会牺牲电池倍率性能,低冰点的添加剂(如乙二醇)将有效提升电池的低温性能,阻燃剂(TMP、TEP)有助于电池安全性,反之,低闪点则尽量避免,以及宽温、低挥发性、低毒和成本等因素124。此外,对于有机溶剂/水电解液调控,研究主要集中在锌负极的保护方面,对正极材料性能的关注较少,因此,更多的有机溶剂添加对正极材料电化学性能的影响还需进一步研究。

表3 常见有机溶剂的物理参数及在水系ZIBs中的作用Table 3 The roles in ZIBs and physical parameters of common organic solvent.
5 总结与展望
通过电解液调控优化策略(有机/无机添加剂、高浓盐、改变溶剂组分等),可以:(1)根据同离子效应,建立电极材料-电解液中活性物质溶解平衡;(2)改变pH值有效裁剪电位平台;(3)通过阴离子或溶剂分子与Zn2+之间强的作用力,破坏Zn2+的溶剂化结构—[Zn(H2O)6]2+,并参与Zn2+溶剂化结构重组,降低溶剂化结构中的水分子数量;(4)破坏水分子间氢键网络结构,降低自由水活性。因此,可以扩宽电化学稳定窗口、抑制正极材料的溶解及界面副反应、在电极材料/电解液界面原位生成CEI保护膜等,有效提升电池的工作电压、容量、维护结构稳定性及延长循环寿命。
然而,已开发的电解液和电极材料之间的普适性及兼容性并不是很高,且添加剂对正极材料性能提升的机制还需更深入的研究和讨论。高浓度电解液本身存在的问题如粘度大、成本高、固-液转化温度高等也需进一步改善。此外,对有机溶剂/水混合电解液主要针对负极侧,很少关注该类电解液体系对正极材料的影响。未来仍需要对电解液体系进行开发和深入挖掘:
(1)电化学机理研究。目前报道的水系ZIBs储能机理主要包括嵌入/脱出机制、转化机制、置换/嵌入机制、溶解-沉积机制等。其中Zn2+/H+共嵌机制是材料获得高容量的重要原因,但同时该机制容易造成电解液局部或整体pH值的增大,伴随在界面生成副产物Zn4SO4(H2O)6·4H2O和(或)Zn2(V2O7)(OH)·nH2O,会增加界面阻抗,阻挡Zn2+在电极材料中的进一步嵌入,还会增加其不可逆性,造成容量的快速衰减,严重影响循环寿命。然而,目前针对副产物的研究较少,尤其是电解液体系和副产物之间的关系方面,其对整个体系电化学性能的影响尚不明确。因此,需通过各种原位表征手段,结合理论计算,深入研究电解液组分、溶剂化结构、分子间氢键等对电化学行为的影响机制。
(2)开发低浓度、宽窗口的电解液体系。电化学稳定窗口一直是制约开发高电压、高能量密度电池的重要因素。高浓度电解液虽然能有效扩宽ESW,但通常伴随牺牲电池的倍率性能,且增加成本。而电解液添加剂方面对ESW的拓宽又涉及溶液pH的变化,这不利于负极锌的长循环。因此,需进一步开发浓度小于5 mol·kg-1的电解液体系。此外,将有机溶剂和水以适当比例混合,能在锌负极形成静电屏蔽层或原位生成SEI,阻止金属锌负极和水的直接接触,且有机分子能破坏Zn2+溶剂化结构以抑制正极材料副反应,破坏水分子间氢键网络结构以降低自由水活性,从而拓宽水系电解液的电化学窗口,能有效结合水系电导率高与有机电解液电化学窗口宽的优点,发展性能优异的水/有机电解液体系具有良好的应用前景。
(3)开发宽温度范围的电解液体系。水系ZIBs受限于电解液水的凝固点和沸点,其温度工作范围较窄,如低温-50 °C,高温和宽温的研究更少,将限制其实际应用。高离液序列的阴离子,在低温性能方面有一定的优势,可以使用Zn(ClO4)2电解液或作为电解液添加剂。此外可以添加低凝固点(如乙二醇)的有机物,和(或)高沸点有机溶剂组分,通过调节水分子间氢键网络结构,可拓宽电池工作温度范围。
