ZnW/β中ZnO含量对其催化四氢萘加氢裂化反应性能的影响
2023-03-14张燕挺陈胜利
王 磊,党 辉,张燕挺,陈胜利
(中国石油大学(北京)化学工程与环境学院,北京 102249)
0 引 言
近年来催化裂化柴油产能过剩已成为常态,而苯、甲苯、二甲苯(benzene/toluene/xylene, BTX)等重要的基础精细化工原料一直处于供不应求状态[1-3],将催化裂化柴油经加氢精制、加氢裂化转化为BTX等轻质芳烃[4],是解决催化裂化柴油产能过剩和缓解轻质芳烃短缺的有效方法[5-7]。而四氢萘(tetralin, THN)及其衍生物(催化裂化柴油加氢精制后产物)加氢裂化是决定BTX等轻质芳烃收率的关键步骤[8]。因此许多研究者以四氢萘等模型化合物作为原料进行加氢裂化反应制备BTX[9-11]。
目前过渡金属和β分子筛因分别具有优异的加氢活性和裂化活性被广泛用作加氢裂化催化剂的加氢和裂化组分。许多研究表明在分子筛处理过程中引入的杂质原子,或覆盖强酸位或与强酸结合形成弱酸(如Zn、Ga)[12-13],可以抑制催化剂的过度裂化性能,使得加氢中心与酸中心得到良好的匹配[14-16],从而提高催化性能。如Jing等[17]研究了Zn、Ga改性的ZSM-5分子筛催化甲醇制芳烃反应。结果表明Zn/ZSM-5能显著提高芳烃收率,当Zn含量为8%(质量分数)时,芳烃收率可达56%,Ga/ZSM-5催化反应的芳烃收率低于Zn/ZSM-5。这归因于Zn的引入可以明显减少强酸量,并且能够抑制裂解和异构化反应,这有利于芳烃的生成。贾艳明等[18]采用等体积超声浸渍法在HZSM-5分子筛上负载Zn、Ga,研究其甲醇制芳烃反应性能。Zn/HZSM-5催化反应具有最高的BTX选择性,Ga/HZSM-5催化反应的BTX选择性较低。作者认为是由于Zn与HZSM-5分子筛的羟基反应生成了ZnOH+,而ZnOH+是一种强脱氢芳构化的Zn-Lewis酸,从而增强了Zn/HZSM-5的脱氢芳构化性能。Shen等[19]研究发现,Zn改性的HY分子筛有利于萘加氢裂化制苯、甲苯、乙苯和二甲苯(benzene/toluene/ethylbenzene/xylene, BTEX)反应,催化反应萘转化率达到98%(质量分数),BTEX选择性达到82%(质量分数),而用Ga改性的HY分子筛催化反应的萘转化率为65%(质量分数),BTEX选择性为65%(质量分数)。作者认为是由于Zn的引入降低了催化剂的酸强度和酸量,抑制了萘的过度裂化,从而提高了BTEX的选择性。安志远等[20]采用等体积浸渍法负载Zn在HZSM-5分子筛上,研究其棕榈油催化生产芳烃性能。在反应温度为500 ℃,Zn负载量为3%时,芳烃收率为59%(质量分数)。作者认为Zn的引入降低了B酸量的同时增加了L酸量,从而增加了芳烃的收率。张长城[21]采用Zn改性的β分子筛作为1-甲基萘加氢裂化制备BTX的催化剂酸组分,结果也表明随着ZnO负载量增加(0~3%,质量分数),BTX收率和BTX选择性均增加,同时生成的积炭量减少。这是由于强酸量会随着Zn含量的增加大幅度减少(Zn与B酸中心反应生成ZnOH+),抑制了1-甲基萘的过度裂化,从而提高了反应活性。
根据本课题组前期的研究结果[22-23],过渡金属W由于有较强的选择性加氢能力而适用于多环芳烃加氢裂化制BTX反应。本文以四氢萘作为催化裂化柴油的模型化合物,以β分子筛为催化剂酸性组分,W为加氢活性组分,研究不同负载量的ZnO对ZnW/β催化剂的理化性质和催化性能的影响。
1 实 验
1.1 实验原料与试剂
H-β分子筛(n(SiO2)/n(Al2O3)=40),南开大学催化剂厂产品;偏钨酸铵([NH4]6W7O24·6H2O,质量分数99.5%),国药集团化学试剂有限公司;硝酸锌(Zn(NO3)2,质量分数98.0%),北京化工厂;二硫化碳(CS2,质量分数99.0%),天津福晨化学试剂厂;四氢萘(C10H12,质量分数97.0%)、环己烷(C6H12,质量分数99.5%),上海麦克林生化科技有限公司。
1.2 催化剂的制备
采用浸渍法将金属氧化物负载于H-β分子筛上。具体制备过程如下:首先称取H-β分子筛平铺于培养皿中,将不同质量分数的硝酸锌溶液逐滴加入到H-β分子筛中,将浸渍后的样品干燥过夜,在马弗炉550 ℃下煅烧3 h。再配制一定质量分数的偏钨酸铵溶液,待其完全溶解后逐滴加入到Zn/β催化剂中,将其在室温条件下干燥过夜,在马弗炉550 ℃下煅烧3 h。最后将上述催化剂压片、筛分后得到直径224~450 μm催化剂样品,分别记为25%W/β、0.5%Zn25%W/β、1%Zn25%W/β、1.5%Zn25%W/β、3%Zn25%W/β、5%Zn25%W/β(质量分数,下同)。
1.3 催化剂的表征
采用Bruker D8 Advance型X射线衍射仪(XRD)对催化剂进行物相分析。测试条件是Cu靶,Kα射线,管电压为40 kV,管电流为50 mA,扫描角度2θ范围为5°~50°。
采用FEI Quanta200F型扫描电子显微镜(SEM)对催化剂的外貌和粒径大小进行分析。催化剂在观察前需要进行喷金处理,喷金采用Leica EMSCD500离子溅射仪,具体时间为150 s,电压为20 kV,电流为19 mA。
采用Thermo Fisher Nicolet Is5型傅里叶红外光谱仪(FT-IR)对催化剂进行分子结构分析,测试波数范围为400~4 000 cm-1。
采用Auto-Micromeritics公司生产的ASAP 2010型比表面及孔隙分析仪(BET)对催化剂的比表面积和体积进行表征。采用BJH法计算催化剂的孔体积,吸附曲线上相对压力为0.05~0.30的数据计算催化剂的比表面积。
采用TPD/TPR 5079型的吸附仪(NH3-TPD)测定催化剂的酸量及酸强度。测试步骤是将500 mg的催化剂样品[20~40目(450~900 μm)]在600 ℃活化30 min,然后110 ℃下吸附氨气30 min; 关闭氨气,基线平稳后以升温速率10 ℃/min将样品加热至600 ℃脱附。
采用AUTO型的Chem 2920自动吸附仪(H2-TPR)测定催化剂的还原能力。测试步骤是将30 mg的催化剂(20~40目)在500 ℃氩气氛围下活化30 min,气流流速为40 mL/min,待冷却至110 ℃后,在保持气流流速不变的条件下,将气体切换为体积分数为6%H2/Ar的混合气,并以10 ℃/min的升温速率升至950 ℃。
采用德国Bruker公司生产的Tensor 27型红外光谱仪测定催化剂的Lewis酸(L酸)、Brønsted酸(B酸)的含量。测试步骤是将20 mg催化剂压成薄片,在1×10-5Pa的真空度下吸附吡啶30 min后,分别程序升温至150、250和350 ℃下进行测试。
采用美国Thermo Fisher公司生产的ESCLAB 250Xi型X射线光电子能谱仪(XPS)对催化剂表面元素化学位移进行测定。条件是辐射源为Al Kα,分辨率为0.5 eV,以污染碳C 1s的结合能(Eb=284.8 eV)为内标。测试步骤为:将催化剂研磨成超细粉体后压片成型,取成型后薄片后进行测试。
1.4 催化剂的加氢裂化反应性能评价
以四氢萘为催化裂化柴油模型化合物,对制备的催化剂进行加氢裂化反应性能评价。在海安石油科研仪器有限公司生产的加氢裂化微型反应装置上进行实验,反应器为内径9 mm、长度500 mm的不锈钢固定床反应器。使用前在反应器中间装入1 g的催化剂颗粒和6 g惰性石英砂颗粒的混合物,反应器的顶部和底部的反应器剩余空间填充20~40目的石英砂。在四氢萘加氢裂化反应之前,需要进行预硫化过程将金属氧化物转化为金属硫化物。催化剂预硫化的条件是氢气的纯度为99.99%,预硫化液为溶有CS2(1.5%,质量分数)的环己烷溶液,反应压力为6.0 MPa,空速为5 h-1,氢油体积比为600∶1。升温过程如下:由室温经过110 min升至240 ℃并保持2 h,再经过30 min升温至320 ℃后保持3 h,再经过20 min升至反应温度400 ℃,进料切换为反应物四氢萘,稳定12 h后,进行加氢裂化反应720 min。反应条件是纯度为99.99%的氢气气氛,反应压力6.0 MPa,温度400 ℃,氢油体积比3 000∶1。
四氢萘的加氢裂化过程反应复杂,反应产物分为气相产物和液相产物,气相产物主要是C1~C4烷烃,采用SP2100 型气相色谱仪进行表征分析,搭载Al2O3毛细柱(30 m×0.530 mm)和氢火焰离子检测器(FID),分析条件是柱箱初温设为80 ℃并保持5 min,再以10 ℃/min的升温速度升温至250 ℃,并保持10 min。液相产物含有C3~C11烷烃,采用SP3420型气相色谱仪分析,色谱柱为PONA毛细柱(50 m×0.2 mm×0.5 μm),检测器为FID。柱箱设为35 ℃并保持15 min,再以2 ℃/min的升温速度升温至250 ℃,并在该温度保持20 min。
四氢萘转化率(XTHN,%)、BTX收率(YBTX,%)和BTX选择性(SBTX,%)定义如下:
(1)
(2)
(3)
式(1)~(3)中:WTHN,0、WTHN,t为四氢萘进料和出料时的质量流速,g/h;WBTX,t为BTX出料时的质量流速,g/h;NTHN,0、NTHN,t为四氢萘进料和出料时的摩尔流速,mol/h;NBTX,t为出料物中BTX的摩尔流速,mol/h;XTHN为四氢萘转化率;YBTX为BTX收率,SBTX为BTX选择性。
2 结果与讨论
2.1 催化剂的表征及分析
2.1.1 催化剂的形貌与结构
图1为不同ZnO负载量催化剂的具有代表性的SEM照片。从图1中可以得出,催化剂颗粒均为不规则球形,球形颗粒小且均匀分散,粒径大约为几百纳米。

图1 不同ZnO负载量催化剂的SEM照片
图2是为不同ZnO负载量催化剂的XRD图谱。从图2中可以得出,H-β分子筛的XRD图谱中2θ在7.8°和22.4°有较好的衍射峰,分别是(101)面和(302)面[24],具有典型的BEA拓扑结构(JCPDS No.47-0183),杂质相可以忽略,结晶度高。25%W/β和ZnW/β催化剂均具有相似的BEA结构特征衍射峰。负载金属氧化物导致衍射峰强度明显下降,并随着金属氧化物负载量的增多,衍射峰强度下降幅度增大,但并未改变和破坏分子筛自身的拓扑结构和晶型。25%W/β和ZnW/β催化剂的XRD图谱2θ在23.0°、23.5°、24.2°和33.7°有明显的WO3特征衍射峰,ZnW/β催化剂的XRD图谱2θ在27.2°和28.7°均出现了ZnO的特征衍射峰,且2θ在30.5°处存在非活性组分ZnWO4晶体的特征衍射峰,这说明WO3和ZnO反应生成了非活性组分ZnWO4晶体。随着ZnO含量的增加,ZnO和ZnWO4的特征衍射峰强度逐渐增大,而WO3的特征衍射峰强度逐渐降低。

图2 不同ZnO负载量催化剂的XRD图谱
图3为不同ZnO负载量催化剂的N2吸附-脱附等温线和孔径分布图。从图3(a)中可以看出,ZnW/β催化剂均在低压(P/P0<0.1)时发生显著吸收,这说明催化剂中含有微孔;随后催化剂的吸附量随着相对压力的增加而增加,当相对压力P/P0达到0.95后达到吸附饱和,这种曲线为典型的I型吸附等温线。从图3(b)可以看出,ZnW/β催化剂的孔隙结构相似,均为典型的微孔型材料,微孔大小均集中在2~10 nm。

图3 不同ZnO负载量催化剂的N2吸附-脱附等温线(a)和孔径分布图(b)
表1为不同ZnO负载量催化剂的表面积及孔体积数据。从表1中可以看出,当在β分子筛上负载ZnO、WO3双金属氧化物后,催化剂的总表面积、微孔表面积及孔体积均减小,这是由于ZnO的负载堵塞了催化剂的微孔孔道,而外表面积随ZnO负载量的增加略有减小[25]。

表1 不同ZnO负载量催化剂的孔道结构性质
2.1.2 催化剂的酸性质
图4为测定不同ZnO负载量催化剂的酸强度和酸量的NH3-TPD图谱。从图4中可以看出,所有催化剂都有两个NH3的脱附峰,将110 ℃至250 ℃处的脱附峰定义为弱酸脱附峰,250 ℃至500 ℃处的脱附峰定义为强酸脱附峰[26]。当在β分子筛上负载ZnO和WO3后,强酸脱附峰均向低温方向移动,这说明ZnO和WO3的引入明显降低了强酸强度。表2是不同ZnO负载量催化剂的酸量。从表2中可以看出,相比H-β分子筛,25%W/β催化剂的强酸量及总酸量明显下降,弱酸量小幅度上升。相比25%W/β催化剂,ZnW/β催化剂的强酸量和总酸量明显下降,这是由于ZnO优先覆盖强酸位点[16],弱酸量先上升后下降,最终酸量下降是因为当ZnO负载量过高时,ZnO会堵塞催化剂的孔道,覆盖部分酸性位,这与吡啶-红外(Py-IR)结果一致。

图4 不同ZnO负载量催化剂的NH3-TPD图谱

表2 不同ZnO负载量催化剂的酸量
图5是为测定不同ZnO负载量催化剂的酸类型和酸量的Py-IR图谱。从图5中可以看出,所有催化剂均在1 450、1 490、1 540、1 610 cm-1处存在吡啶分子的脱附峰,其中波数为1 540 cm-1的吸收峰是Brønsted酸中心作用的吡啶分子特征吸收峰,波数为1 450、1 610 cm-1的吸收峰是Lewis酸中心作用的吡啶分子特征吸收峰,波数为1 490 cm-1的吸收峰是Brønsted与Lewis酸中心共同作用的吡啶分子特征吸收峰[27]。表3是在脱附温度为350 ℃下ZnW/β催化剂的Brønsted酸量、Lewis酸量及总酸量的计算结果。从表3中可以看出,相比25%W/β催化剂,随着ZnO含量的增加,ZnW/β催化剂的Brønsted酸量、Lewis酸量及总酸量逐渐下降。这是因为ZnO堵塞了催化剂的孔道,覆盖了大部分酸性位,导致酸量的下降。对于四氢萘的加氢裂化反应,催化剂的酸量过大会容易产生积炭,从而降低反应活性,而催化剂的酸量过小会降低四氢萘的裂化程度,所以加氢裂化催化剂需要具有适中的酸量。引入适量的ZnO可以降低催化剂的酸量,避免四氢萘过度裂化,同时减少积炭的发生。

表3 不同ZnO负载量催化剂的B酸位和L酸位密度(吡啶脱附温度:350 ℃)

图5 不同ZnO负载量催化剂的吡啶-红外图谱
2.1.3 催化剂的金属状态、官能团和还原能力的表征
图6是为研究负载金属的氧化状态所测定的催化剂W 4f的XPS图。从图6可以看出,不同ZnO负载量的催化剂W 4f的XPS分为两个峰,分别是在36.15 eV处低电子结合能的主峰(W 4f7/2)和在38.20 eV处高电子结合能的主峰(W 4f5/2)[28]。同时,对比各个催化剂的XPS位置上的变化可以发现,在25%W/β催化剂中引入ZnO后,W 4f5/2和W 4f7/2峰均向低电子结合能方向偏移,这是由于ZnO的引入使得WO3与β分子筛之间的相互作用力不断减弱。WO3与β分子筛的相互作用力:[25%W/β]>[0.5%Zn25%W/β]>[1%Zn25%W/β]>[1.5%Zn25%W/β]。
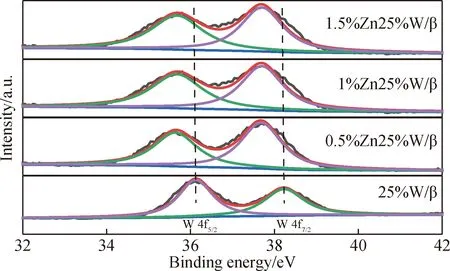
图6 不同ZnO负载量催化剂W 4f的XPS
图7是为研究不同ZnO负载量催化剂的官能团所测定的FT-IR图谱。从图7中可以看出,25%W/β、0.5%Zn25%W/β、1%Zn25%W/β与5%Zn25%W/β催化剂的FT-IR图谱均在462、575、810 、1 096、1 230、1 630、3 440 cm-1处有峰。其中462和1 096 cm-1处的峰是β分子筛中Si—O—Si的弯曲振动峰和反对称伸缩振动峰,1 230 cm-1处的峰是β分子筛中T—O—T(其中T是Si或者Al)反对称伸缩振动峰[29],1 630和3 440 cm-1处的峰是β分子筛中Si—O(H)—Al中羟基的弯曲振动峰和伸缩振动峰[30],575和810 cm-1处的峰是WO3中W—O键的反对称弯曲振动峰和对称伸缩振动峰[31]。另外,1%Zn25%W/β和5%Zn25%W/β催化剂的FT-IR图谱均在1 390 cm-1处有峰。这是当ZnO含量达到1%后,WO3和ZnO产生较强的相互作用形成Zn—O—W键而产生的峰,且随着ZnO负载量的增加,Zn—O—W键的峰强度也增大。

图7 不同ZnO负载量催化剂的FT-IR图谱
图8是为研究金属氧化物与载体之间的相互作用所测定的H2-TPR图谱。从图8中可以看出,25%W/β有两个还原峰,其中723 ℃处是八面体WO3的还原峰,853 ℃处是四面体WO3的还原峰[24]。0.5%Zn25%W/β有两个还原峰,其中708 ℃处是八面体WO3的还原峰,815 ℃处是四面体WO3的还原峰。1%Zn25%W/β催化剂有三个还原峰,其中690 ℃处的峰是八面体WO3的还原峰,796 ℃处的峰是四面体WO3的还原峰,887 ℃处是ZnWO4晶体的还原峰[32-33]。5%Zn25%W/β催化剂有三个还原峰,655 ℃处是八面体WO3的还原峰,791 ℃处是四面体WO3的还原峰,910 ℃处是ZnWO4晶体的还原峰。相比25%W/β催化剂而言,随着ZnO负载量的增加,ZnW/β催化剂的八面体WO3的低温氢气还原峰和四面体WO3的高温氢气还原峰进一步向低温处移动。说明催化剂中引入ZnO后,WO3与β分子筛之间的相互作用力逐渐减弱。这是由于ZnO与β分子筛作用力大于ZnO与WO3作用力,导致ZnO与β分子筛之间的作用代替了WO3与β分子筛之间的作用,使WO3聚集,提高了WO3的还原能力,这与XPS结果一致。1%Zn25%W/β催化剂峰值在887 ℃处的还原峰与5%Zn25%W/β催化剂峰值在910 ℃处的还原峰均是非活性组分ZnWO4晶体的还原峰。这是由于覆盖在β分子筛外表面的ZnO含量达到1%后,ZnO与WO3会反应生成难还原的ZnWO4晶体[34],且ZnWO4晶体的还原温度随ZnO的含量增加而增加,这与XRD结果一致。对比1%Zn25%W/β催化剂和5%Zn25%W/β催化剂的WO3还原峰和ZnWO4晶体还原峰,可以发现随着ZnO负载量的增大,非活性组分ZnWO4晶体的生成量增大,非活性组分ZnWO4晶体与WO3的比例逐渐增大,使得WO3在金属氧化物中的比例逐渐减小,从而导致ZnW/β催化剂的加氢中心与酸中心匹配不佳。

图8 不同ZnO负载量催化剂的H2-TPR图谱
2.2 不同ZnO负载量的ZnW/β催化四氢萘加氢裂化制BTX的反应性能
以四氢萘为原料,分别以25%W/β、0.5%Zn25%W/β、1%Zn25%W/β、1.5%Zn25%W/β、3%Zn25%W/β、5%Zn25%W/β为加氢裂化催化剂,在反应温度为400 ℃、反应压力为6 MPa和氢油体积比为3 000∶1的反应条件下对不同ZnO负载量的ZnW/β催化剂进行加氢裂化反应性能评价。反应均在催化剂运行稳定期进行测试评价。
图9为不同ZnO负载量的ZnW/β催化剂催化四氢萘加氢裂化制备BTX反应的转化率、气相收率、BTX收率随空时的变化及BTX选择性随转化率的变化图。从图9(a)中可以看出,随着空时的提高,四氢萘的转化率均逐渐增大,25%W/β催化四氢萘的转化率比ZnW/β的高,这是由于25%W/β催化剂的酸强度比ZnW/β催化剂高,裂化能力强。对比不同ZnO负载量的ZnW/β催化四氢萘的转化率,可以发现随着ZnO含量的增加,四氢萘的转化率逐渐降低,这是由于催化剂的酸强度逐渐降低,裂化能力变弱。从图9(b)中可以看出,随着空时的提高,气相收率均逐渐增大,25%W/β催化四氢萘的气相收率比ZnW/β的高,这是由于25%W/β催化剂的酸强度较高导致四氢萘过度裂化为气体。对比不同ZnO负载量的ZnW/β催化四氢萘的气相收率,发现随着ZnO含量的增加,气相收率逐渐降低,这也是催化剂酸强度逐渐降低,裂化能力变弱导致的。从图9(c)中可以看出,所有催化剂催化四氢萘制得的BTX收率随着反应空时的提高均呈现出先增大后减少的火山型趋势。与其他催化剂相比,1%Zn25%W/β催化剂的催化反应具有最高的BTX收率,在反应空时0.36 h时,BTX收率最高达到41.57%(质量分数)。从图9(d)中可以看出,所有催化剂催化四氢萘制得的BTX选择性随着转化率的增加均呈现出先增大后减少的火山型趋势。与其他催化剂相比,1%Zn25%W/β催化剂的催化反应具有最高的BTX选择性,在转化率为94.54%时,BTX选择性最高达到44.1%(质量分数)。对比不同ZnO负载量的催化剂的催化反应的最高BTX收率及最高BTX选择性,可以发现[1%Zn25%W/β]>[1.5%Zn25%W/β]>[0.5%Zn25%W/β]>[25%W/β]>[3%Zn25%W/β]>[5%Zn25%W/β]。

图9 不同ZnO负载量的催化剂加氢裂化催化性能
图10是不同ZnO负载量的催化剂催化反应的最高BTX选择性。从图10中可以看出,随着ZnO负载量的增大,最高BTX选择性先增大后减小。这是由于负载ZnO可以适当降低催化剂的酸强度,抑制四氢萘的过度裂化,从而提高BTX的选择性。而当ZnO的负载量达到1%后,ZnO和WO3反应生成非活性组分ZnWO4,并随着ZnO负载量的增加,非活性组分ZnWO4晶体的量逐渐增大,非活性组分ZnWO4晶体与WO3的比例逐渐增大,WO3在金属氧化物中的比例逐渐变小,使得酸中心与加氢中心匹配不佳,从而导致ZnW/β催化反应的最高BTX选择性随着ZnO负载量的增加先升高后降低。相比其他催化剂,1%Zn25%W/β催化剂的最高BTX选择性最大,说明该催化剂的酸量适中且加氢中心与酸中心匹配最佳。

图10 不同ZnO负载量的催化剂催化反应的最高BTX选择性
表4为负载不同ZnO含量催化剂反应后的积炭含量(wcoke)。从表4中可以看出,相比25%W/β催化剂,ZnW/β催化剂的积炭含量随着ZnO含量的增加逐渐减小,催化剂的积炭含量下降是由于积炭的生成主要在β分子筛酸性中心上,ZnO的引入使β分子筛的比例下降,导致强酸量和总酸量的降低,积炭含量明显下降。可见在催化剂中引入适量的ZnO可以有效提高催化剂的抗积炭能力。

表4 不同ZnO负载量的催化剂催化反应的积炭含量
3 结 论
采用浸渍法负载不同含量的ZnO和一定量的WO3在β分子筛上制成ZnW/β加氢裂化双功能催化剂。结果表明,ZnW/β的催化反应最高BTX收率随着ZnO负载量的增加呈现先升高后降低的趋势。BTX收率升高是由于ZnW/β催化剂的强酸量和总酸量随着ZnO含量的增加明显下降,抑制了四氢萘的过度裂化。BTX收率降低是当ZnO含量达到1%后,ZnO与WO3反应生成的非活性组分ZnWO4晶体随着ZnO负载量的增大而增大,降低了催化剂中加氢活性组分WO3的含量,导致ZnW/β催化剂的加氢中心与酸中心匹配不佳。1%Zn25%W/β催化反应在反应空时为0.36 h时,表现出最高的BTX收率(41.57%)及最高的BTX选择性(44.1%),说明该催化剂的酸量适中且加氢中心与酸中心匹配最佳。迄今为止,研究者们普遍认为ZnO助剂在加氢裂化催化剂中的作用是调变催化剂的酸量。本研究工作发现ZnO会与WO3反应生成ZnWO4晶体,降低了催化剂中加氢活性组分WO3的含量,影响催化剂加氢中心与酸中心的匹配程度。
