以发热为首发症状的组织细胞坏死性淋巴结炎1例及文献复习
2023-03-11胡燕灵纪娟张念志
胡燕灵 纪娟 张念志
组织细胞坏死性淋巴结炎(histiocytic necrotizing lymphadenitis,HNL)是一种以非肿瘤性淋巴结反应性增大为特征的疾病,具有自限性,根据文献报道及分析,多见于中国、日本等东南亚国家40岁以下青壮年患者,其中又以女性多见[1],属于临床罕见疾病,容易被误诊为转移瘤、恶性淋巴瘤等疾患。本文通过分析临床学习过程中所见1例HNL患者的临床诊疗过程,同时结合文献进行学习,对其发病机制、临床表现及诊断依据等进行探讨,以提高对本病的认识,也为不明原因发热患者的诊断提供思路。
临床资料
患者,男,33岁,因“发热1周”于2021年2月4日由门诊拟“发热待查”收住呼吸内科。患者自诉1周前受凉后出现发热,最高体温达39℃,晨起及夜间持续发热,恶寒,少许咳嗽、咳痰,咳白痰,伴有颈部疼痛,全身酸痛、乏力。曾于2021年1月30日就诊于外院,查CRP、血常规、胸部CT均未见明显异常。予“奥司他韦75mg bid 及莲花清瘟胶囊4粒tid”口服治疗,症状改善不明显。2021年2月4日为求进一步治疗,就诊我院呼吸内科,门诊拟“发热待查”收住入院。
查体:患者入院时测体温:38.2℃,神清,精神欠佳,咽部无充血,扁桃体无肿大,扁桃体表面无脓性分泌物,左侧颈项部触及少许黄豆般大小增大淋巴结,压痛(+),质软,活动度一般,听诊双肺呼吸音粗,未闻及明显干湿性啰音,心率116次/分,律齐,腹软,无压痛及反跳痛,双下肢不肿。
诊疗经过:入院完善相关检查,2021年2月4日急诊血常规:白细胞计数4.19×109/L,红细胞计数5.62×1012/L,血红蛋白164g/L,血小板计数178×109/L,中性粒细胞计数2.76×109/L,中性粒细胞百分比65.9%,淋巴细胞计数1.14×109/L,淋巴细胞百分比 27.2%;2021年2月5日尿常规:尿蛋白 弱阳性,白细胞56.3个/uL↑,上皮细胞 91.20个uL↑,小圆上皮细胞 84.40个/uL↑;凝血常规:纤维蛋白原5.16 g/L↑,D-二聚体2.7 mg/L↑;生化:ALT 58 U/L↑,AST 42 U/L↑,总蛋白61.9g/L↓,白蛋白35.7 g/L↓,乳酸脱氢酶419 U/L↑,CRP:10.48 mg/L↑,HsCRP:9.80 mg/L↑;呼吸道病原体谱:抗流感病毒A型-IgM 弱阳性,抗流感病毒B型-IgM 弱阳性;自身抗体:抗nRNP/Sm抗体 临界阳性(±),抗Sm抗体 临界阳性(±),肿瘤标志物:铁蛋白 1650.91 ng/mL↑;2021年2月8日肥达氏反应:肥达氏0效价1 ∶80(+),肥达氏H效价1 ∶20(+);心肺功能五联定量测定:D-二聚体1860 ng/mL↑;余大便常规、真菌D-葡萄糖检测、优生优育系列、降钙素原、甲状腺全套未见明显异常。2021年2月5日查胸部CT及全腹部CT均未见明显异常。治疗上西医予维生素C 2g+维生素B60.2g 静滴 qd补液、甲磺酸左氧氟沙星 0.5g qd抗感染、痰热清 20mL qd退热等对症治疗;中医予疏风清热,解毒生津之剂口服,效果不明显,患者仍发热。2月4日-2月6日患者反复高热,体温最高可达39.3℃。
患者发热病因不明,2月6日完善风湿科、血液科会诊后予行浅表淋巴结彩超及相关检查,以明确发热病因。2月7日上午真菌(1-3)- β-D葡聚糖<10pg/mL;浅表淋巴结彩超示:左侧颈部多发淋巴结,较大淋巴结建议超声引导下穿刺活检;呼吸道病原体谱提示:抗流感病毒A型-IgG阳性(±),抗流感病毒b型-IgM弱阳性(±),遂予奥司他韦75mg bid口服。考虑患者使用左氧氟沙星抗感染治疗3天仍有发热,予以停用,调整为哌拉西林舒巴坦4.5g bid继续抗感染。结合患者淋巴结彩超结果,考虑组织细胞坏死性淋巴结炎可能,遂与患者及家属沟通后予完善淋巴结活检病理检查,同时加用甲泼尼龙琥珀酸钠40mg qd静滴抗炎退热,2月7日下午复测体温正常,后未再发热。2月9日患者诉颈部疼痛明显好转,体温正常,一般情况可,淋巴结穿刺病理、T-spot、血培养结果尚未回示,患者及其家属要求出院,子以办理。嘱其以泼尼松初始剂量20mg bid 口服(每5天减5mg)巩固治疗。2月10日患者病理回示:淋巴组织多灶及片状坏死,内见多量核尘,散在组织细胞、淋巴细胞、免疫母细胞及少量浆细胞样细胞浸润,局部副皮质区细胞增生,考虑诊断为组织细胞坏死性淋巴结炎(Kikuchi病,如图1)。1月后患者于我科门诊随访,调整用药,复查浅表淋巴结彩超未见明显异常。
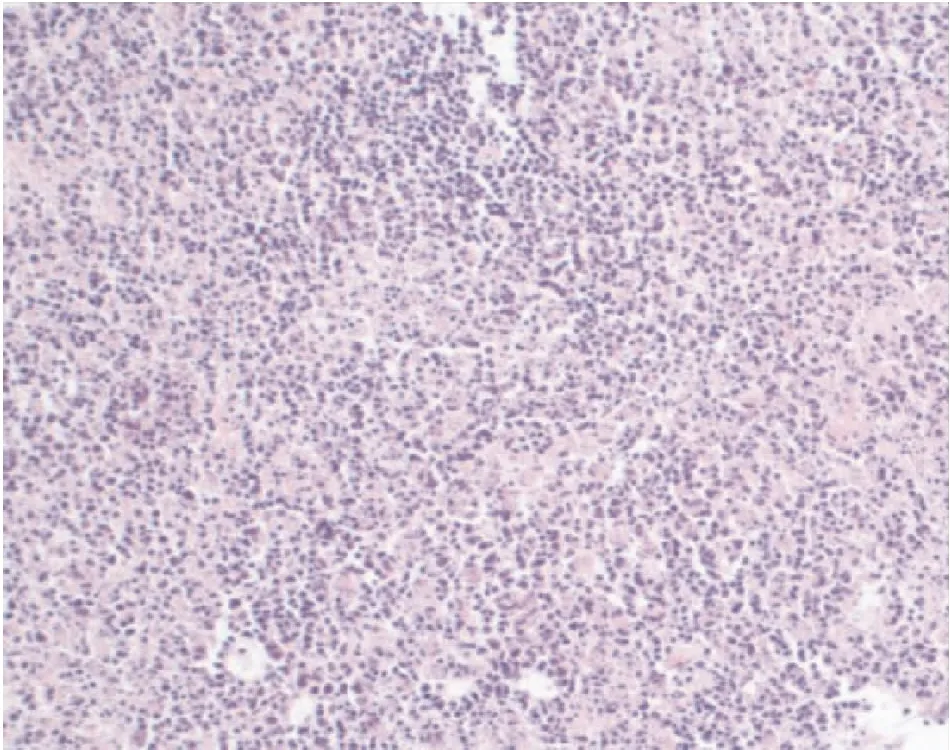
图1 淋巴结病理结果(HE,sp×200)
讨 论
病因及发病机制:HNL最早于1972年由日本学者Kikuchi及Fujimoto提出,故又称Kikuchi病或Kikuchi-Fujimoto病[2-3],是一种临床罕见疾病,其发病原因尚不明确,但其发热、淋巴结肿大、白细胞一过性减少、抗生素治疗无效及其发病前上呼吸道感染的前驱症状等临床特征,提示其发病可能与病毒感染后所诱发的高免疫状态相关,病毒感染包括人疱疹病毒6型和8型、EB病毒、巨细胞病毒、副流感病毒、微小病毒B19、柯萨奇病毒、人类免疫缺陷病毒等[4-5],HNL患者在病毒入侵机体后,产生过度免疫反应,从而破坏机体免疫平衡,最终淋巴细胞变性坏死,被组织细胞吞噬降解而发生此病。Moyer等学者[6-7]的研究表明,EB病毒富集于人体淋巴结区,干扰T淋巴细胞介导的正常免疫应答反应,又或者直接侵犯淋巴组织而致病。Yosep Chong等[8]运用定量聚合酶链反应(qPCR)技术证实Torque-teno病毒/Torque-tenolike迷你病毒在HNL的发病机制中具有重要意义,可能是发病原因之一。本例患者发病前有受凉病史,存在恶寒发热,咳嗽咳痰,全身乏力酸痛等上呼吸道感染症状,呼吸道病原体谱示流感病毒A型、B型IgM抗体弱阳性,与此发病机制相符,推测该患者发病过程可能是由于病毒感染引起自身免疫反应失衡,诱导淋巴细胞变性坏死。但也有相关研究指出[9]:在对20例HNL患者组织标本的PCR检测中,仅有2例标本的个别细胞中检测出少量的EB病毒信号,其阳性检测率仅10.0%,无统计学意义。也有其它研究人员[10]提出HNL的发病不仅与病毒相关,病原体还包括常见的感染菌如布鲁氏菌、耶尔森氏菌、弓形虫等。此外,临床研究发现,本病的发生还与自身免疫系统疾病存在一定关联,Perry等[11-12]曾报道HNL与系统性红斑狼疮等免疫系统疾病存在一定相关性,两者可并发或少数HNL患者可进展为SLE。但无论是病毒感染或是自身免疫因素,其在HNL疾病中的具体作用机制尚未能得到有力的证据支持,有待于进一步研究。
临床特征及诊断依据:HNL临床表现复杂多样,典型临床特征[10]包括长期不明原因发热,或持续高热,或低热,且热型不一;淋巴结肿大,多见于颈后三角区的淋巴结肿大或伴疼痛,也可见于腋窝、锁骨上窝、腹腔、腹股沟等部位的淋巴结,肿大的淋巴结多呈串珠样排列,直径可介于0.5~4cm之间,少数见于5~6cm,极少数超过6cm。其它临床症状还包括头痛、寒战、盗汗、恶心、食欲下降、疲乏、呼吸困难、全身皮疹、关节肌肉疼痛等,非典型症状起病时易与其它疾病相混淆,误诊率高。实验室检查显示有60%HNL患者可有白细胞一过性减少,中性粒细胞减少时意义较大,部分患者可有贫血,血小板减少症,红细胞沉降率(ESR)加快,C-反应蛋白(CRP)升高,乳酸脱氢酶及肝酶的升高等,血清中或可检测到EB病毒、巨细胞病毒、人疱疹病毒等的相关抗体,有长期发热及头疼等神经系统症状时可行脑脊液的检查,实验室检查指标缺乏特异性,难以确诊[13]。常规影像学检查主要包括B超及CT,亦缺乏特异性,B超检查常表现为单侧颈部淋巴结肿大,多见于Ⅱ区和V区,病变的淋巴结直径通常小于3cm,有研究将[14]B超下的肿大淋巴结特点概括为低回声坏死、强回声钙化少、圆形及椭圆形、淋巴结门结构及其边界不清。亦有研究表明[15]淋巴结周围软组织呈高回声晕是HNL彩超下的较为显著的特征,这一特征有助于本病与其它淋巴结疾病相鉴别,但也可见于淋巴结结核、感染性淋巴结炎等[16],故虽对HNL诊断具有一定意义,仍缺乏特异性。超声造影提示[17]HNL虽与恶性淋巴结病变有所交叉,但其造影表现更符合离心性和弥漫性增强的良性淋巴结病变,这为临床明确诊断HNL提供有效参考信息。普通CT特异性不高,常表现为多发微小肿大淋巴结,直径一般小于2.5cm[18],可为临床医生结合患者临床表现及实验室检查结果,与结核性淋巴结炎、恶性淋巴瘤等疾病相鉴别,做出考虑为HNL诊断的可能性,制定下一步治疗计划提供参考依据,但仅凭影像学表现难以确诊HNL。由于HNL早期也常被误诊为转移瘤、恶性淋巴瘤等,18F-FDG PET-CT的应用也较为广泛,在成像中[19-20]多显示为全身多部位的淋巴结轻中度增大,颈部及腋窝淋巴结最常受累,受累淋巴结密度均匀无融合,FDG摄取值增高,或伴中轴骨代谢升高及肝脾肿大。18F-FDG PET-CT在评估HNL患者全身淋巴结累及情况,辅助淋巴结活检定位等方面具有一定意义。虽然18F-FDG PET-CT灵敏度很高,但其在肉芽肿性疾病、炎性疾病等良性病变中也存在类似的影像学表现,特异性较低,且费用高昂,辐射性大,因此对于不明原因发热伴淋巴结肿大患者,18F-FDG PET-CT并非首选,应结合患者临床表现,实验室及B超检查,尽早行活检明确病理检查结果。典型的HNL病理学表现[21]为镜下可见副皮质区淋巴结结构丧失,出现大片或小灶的凝固性坏死,其内可见大量核细胞碎片,坏死边缘见增生的组织细胞及其被吞噬的细胞核碎片,病灶中淋巴细胞呈散在的灶状分布,且掺杂有增生的免疫母细胞,而中性粒细胞及浆细胞浸润较少或无,其中,本病的特征性病理改变之一是病灶区出现碎屑样坏死。既往报道[1,16]曾提及将HNL病理学表现分为增生型、坏死型、黄瘤样型,增生型主要表现为病灶区出现增生的组织细胞、免疫母细胞、核细胞碎片、小淋巴细胞等,但无明显坏死及中性粒细胞的浸润;坏死型则是在增生型基础上发生明显的凝固性坏死,并包含较多的核碎片;黄瘤样型则是泡沫样组织占据整个病变区域。阐明了HNL进行性发展过程中的阶段性病理改变。非典型者可在镜检基础上进行免疫细胞化学检查,相关临床研究表明[9]病灶细胞CD3、CD4/CD8、MPO、CD123、颗粒酶B及TIA1阳性,CD20、CD56、PAX5阴性对于HNL的诊断及鉴别诊断具有一定意义。本例患者后续镜检结果中描述的病理特征与文献报道一致,诊断明确,因而在临床诊疗过程中,对于不明原因发热,尤其伴有淋巴结肿大的患者,需考虑到本病与其它类似疾病的可能性,有适应证者尽早安排活检,明确病理诊断,针对性治疗,减少误诊漏诊。
治疗及预后:HNL是一种良性自限性疾病,通常在4~6个月之间可自行恢复,预后良好,少数患者(约3%~4%)可出现病情反复或进展为其它疾病,如自身免疫性肝炎、SLE、结缔组织病等,Chen Z.Y.等[22]曾报道1例HNL患者经糖皮质激素治疗好转后出院,随访2月出现肝功能损害,后诊断为自身免疫性肝炎(AIH)继续入院治疗的;Vithoosan S.、Lee S.M.[13、23]分别报道1例经病理诊断为HNL,但继发嗜血综合征(HLH)的患者。因而对于HNL也需要长期随访,以预防其复发或进展为其它疾病。在治疗上,临床以对症治疗为主,有发热症状的患者早期可适当运用解热镇痛药物,神经系统受累者,需对症控制颅压及惊厥等。由于本病临床罕见,通常发热患者治疗初期会运用抗生素对症处理,结果抗感染治疗常无效,目前文献报道该病对糖皮质激素治疗敏感[24],但应注意避免在活检前使用。多数患者经糖皮质激素治疗后症状迅速缓解,少数病情严重患者可运用类固醇激素和羟氯喹进行治疗,病情更甚者静脉使用免疫球蛋白效果较好[21,25]。曾有报道显示[26]糖皮质激素效果不明显或病情反复的HNL患者改用羟氯喹或免疫球蛋白治疗后,症状明显缓解。目前国内专家学者[27]多倾向于尽早使用糖皮质激素,推荐泼尼松用量为0.5mg/kg,疗程持续4~6周,由于本病存在3%~4%的复发率,可能与过早停用糖皮质激素相关,故少数患者需延长使用激素至3个月以上。国外研究者则认为,该病多数不需要处理,而仅有大约16%的病人需要糖皮质激素或免疫抑制剂加以治疗。本例患者以发热为首发症状,伴随上呼吸道感染症状,在疾病初期,使用抗生素进行处理无效,考虑诊断为HNL后应用甲泼尼龙琥珀酸钠40mg qd静滴治疗,后体温迅速恢复正常,症状也逐渐好转,出院后以泼尼松20mg bid 口服(每5天减5mg)巩固治疗,定期随访,调整用药,未再复发。
小 结
HNL虽然临床罕见,但在临床诊疗过程中,若遇不明原因发热,淋巴结无痛或痛性肿大,白细胞一过性减少,或伴有皮疹、关节疼痛、贫血、肝脾肿大等不典型症状的患者,需要警惕该病发生的可能性,有适应证者应尽早活检,明确病理诊断,及早治疗,以免漏诊误诊,贻误病情,同时注意与其它疾病相鉴别,病情稳定后,也需要加强定期随访,监测病情,预防疾病进展。
