一例马凡综合征家系患者的临床表现及遗传学分析
2023-03-09孔志华郭俊王月丽李小燕张晓萍陈丽
孔志华,郭俊,王月丽,李小燕,张晓萍,陈丽
马凡综合征由法国医师Antonie Marfan 于1896年首次报道,是一种常染色体显性遗传病;无种族特异性[1]。其可累及心血管、眼部及骨骼等多个系统。主要临床表现为蜘蛛指/趾、脊柱侧弯、主动脉病变、瓣膜病变、晶状体脱位等[2]。人原纤维蛋白1基因(FBN1)是目前已经明确的马凡综合征的致病基因,包含66 个外显子,位于染色体15q21.1 区,编码原纤维蛋白-1[3]。原纤维蛋白-1 通过参与形成微纤维,在维持细胞外基质稳定性中起着重要作用[4]。目 前 为 止,ClinVar(https://www.clinicalgenome.org/data-sharing/clinvar/)数据库中包含超过2 000 种马凡综合征相关的FBN1突变,研究表明FBN1突变无明显热点突变[5-6]。近期研究显示,患者携带的突变类型与临床表型有一定关联,通过分析马凡综合征患者的FBN1基因突变,有助于辅助临床诊断及治疗决策;《遗传性胸主动脉瘤/夹层基因检测及临床诊疗专家共识》阐述了基因检测在遗传性胸主动脉病诊断及筛查中的作用,并明确针对各种类型胸主动脉病患者提出医疗和生活方式管理方面的建议,从而实现对高危人群的早诊断及早预防[7]。本研究通过对1 例临床表现为主动脉夹层的马凡综合征患者进行全外显子测序,经过生物信息学分析,鉴定出一疑似致病突变,并在家系内对该位点进行验证,探讨了该马凡综合征家系的致病基因及致病突变,并为该家系相关成员的临床诊断及进一步诊疗提供了理论依据。
1 资料与方法
1.1 研究对象
选取2019 年6 月因主动脉夹层于首都医科大学附属北京安贞医院心外科就诊的1 例26 岁女性马凡综合征患者(先证者)及其家属为研究对象,患者家系图见图1。经影像学检查(超声心动图、主动脉CT 血管造影、正位X 线胸片及彩色多普勒血管超声)及相关辅助检查,并依据2017 年中国医师协会心血管外科分会发布的主动脉夹层诊断与治疗规范中国专家共识[8]和欧洲心脏病学会发布的2014 年主动脉疾病诊疗指南诊断标准[9],该患者临床诊断为马凡综合征。本研究遵循《赫尔辛基宣言》,通过了首都医科大学附属北京安贞医院伦理委员会批准(批准函编号:2016017),患者及家系成员均签署了知情同意书。
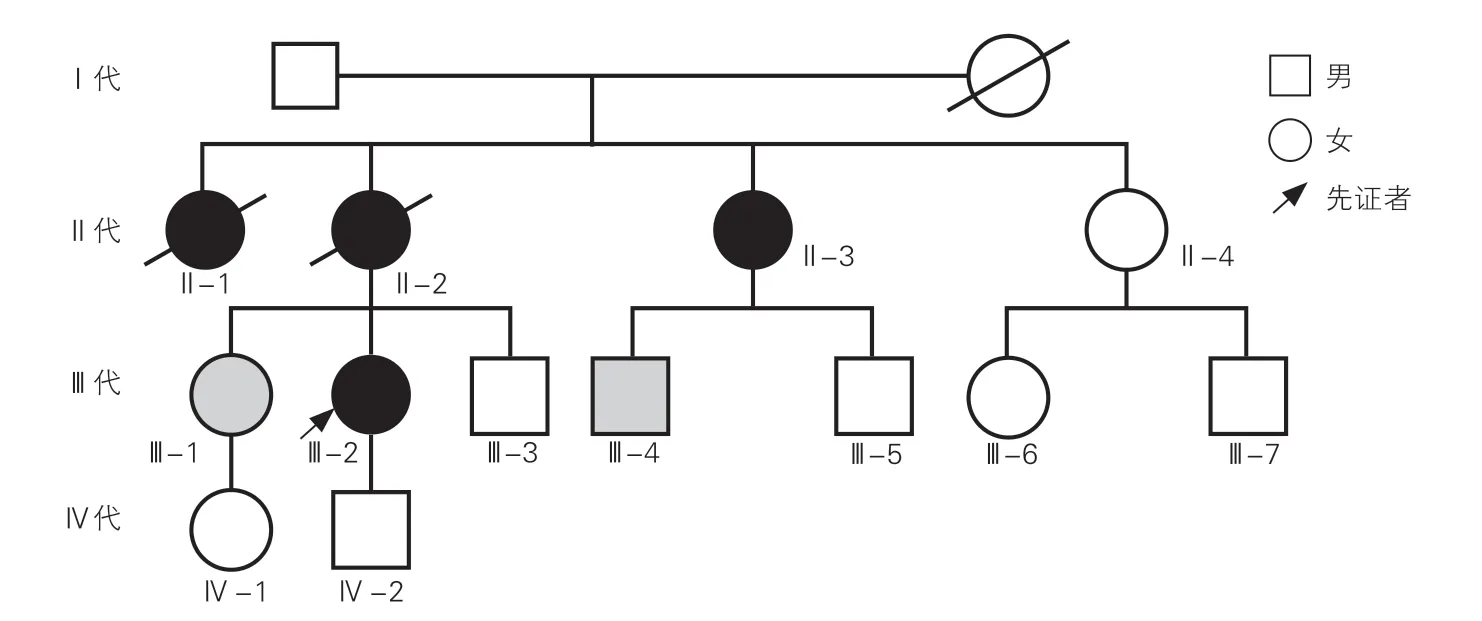
图1 患者家系图
1.2 方法
1.2.1基因组DNA 提取
采集先证者及所有入组家系成员外周血各2 ml[乙二胺四乙酸(EDTA)抗凝];每个样本取200 μl 血样使用全血DNA 提取试剂盒(康为世纪,江苏泰州)提取基因组DNA,利用琼脂糖凝胶电泳对DNA 提取质量进行检测。
1.2.2全外显子测序
基因组DNA 经过质控后,取1 μg 用于文库构建;文库构建使用NEB Next Ultra DNA Library Prep Kit for Illumina(NEB 公司,美国),外显子区域富集使用Agilent SureSelect Human All Exon V6 试剂盒(安捷伦公司,美国),文库构建完成后,先使用Qubit 2.0 进行初步定量,使用实时荧光定量PCR 方法对文库的有效浓度进行准确定量,以保证文库质量。使用Novaseq 进行高通量测序,将通过质控的测序数据比对至人类参考基因组(GRCh37/hg19),平均测序深度大于200 Ⅹ,检测样本的变异信息。利用ANNOVAR 对变异信息进行注释,包括在dbSNP 数据库、千人基因组、本团队内部数据库等数据库的频率信息,同时对变异的位置信息、类型、保守性等进行预测。
1.2.3Sanger 测序验证
利用ABI 3730xl 测序仪(Applied Biosystems 公司,美国)对高通量测序得到的候选位点进行检测。PCR扩增使用引物如下:正向:GGGCAATGGTCAATTCTA,反向:GTGTCTATTTCTGATGGCTTAT,测序使用正向引物。
1.2.4功能预测及变异的临床注释
使用ANNOVAR 软件包Mutation taster(http://www.mutationtaster.org/)、phyloP46way_placenta、Swiss-model(https://swissmodel.expasy.org)等对变异位点进行功能及保守性分析;根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南及中国2017 版《遗传变异分类标准与指南》对基因变异进行分类[10]。
2 结果
2.1 临床特点
先证者(Ⅲ-2)为26 岁女性,主诉“体检发现降主动脉夹层2 个月”,无明显胸、背及腹部疼痛,于当地医院诊断为主动脉夹层。入院查体:脉搏70 次/min,血压125/85 mmHg(1 mmHg=0.133 kPa),呼吸15 次/min,体温37 ℃,身高170 cm,体重53 kg,身材瘦长、蜘蛛指、漏斗胸、关节活动度大、高度近视并伴有皮纹等马凡综合征相关体征与症状[1]。其母亲(Ⅱ-2)及姨妈(Ⅱ-1)因急性主动脉夹层去世,姨妈(Ⅱ-3)因主动脉夹层(A 型)/动脉瘤行主动脉置换术;姐姐(Ⅲ-1)及表弟(Ⅲ-4)超声心动图结果提示主动脉根部增宽;其余家族成员超声心动图未见明显异常。
先证者超声心动图提示主动脉窦部增宽39 mm,三尖瓣少量反流,各心腔内径处于正常范围,各室壁厚度及运动正常(图2)。主动脉CT 血管造影提示主动脉夹层,累及右侧肾动脉及髂总动脉(图3、4)。诊断为主动脉夹层形成、胸主动脉瘤。

图2 先证者超声心动图结果

图3 先证者主动脉CT 成像图

图4 先证者主动脉CT 二维成像图
患者后期接受全胸、腹主动脉置换术,预后较好。
2.2 全外显子测序结果
先证者全外显子测序共得到13.83 GB 有效测序数据,与参考基因组比对后得到14 909 个插入/缺失多态性,126 539 个单核苷酸多态性;依据《遗传性胸主动脉瘤/夹层基因检测及临床诊疗专家共识》建议,筛选与综合征型胸主动脉瘤/夹层相关的FBN1、TGFBR1、TGFBR2、SMAD3、TGFB2、TGFB3、SMAD2共7 个基因的变异位点[7],检测到FBN1基因一杂合变异:FBN1,NM_000138.5:c.7412delC,位于基因第60 号外显子上,为移码突变(图5)。其余6 个主动脉疾病相关基因未检出变异位点。

图5 变异位点c.7412delC 在FBN1 基因上位置示意图
2.3 Sanger 测序结果
先证者的姨妈(Ⅱ-3)、姐姐(Ⅲ-1)、表弟(Ⅲ-4)均检测出与先证者(Ⅲ-2)相同的移码突变(FBN1,c.7412delC),而该家系中无临床表型的健康者,包括先证者姨妈(Ⅱ-4)、弟弟(Ⅲ-3)、两位表弟(Ⅲ-5、Ⅲ-7)、表妹(Ⅲ-6)及先证者之子(Ⅳ-2)及其姐姐之女(Ⅳ-1)均未携带该变异位点(图6)。

图6 Sanger 测序结果
2.4 变异位点功能预测及临床注释结果
该位点在dbSNP、ExAC、ESP6500 等数据库中均未见频率报道,在正常人群中发生的概率极低,提示该变异不是常见良性变异。生物信息学软件预测结果显示,该位点为第60 号外显子区域单个碱基的缺失,该变异导致第2 471 位以后的氨基酸发生了移码变异,造成翻译的提前终止,组成蛋白质的氨基酸为2 681 个(野生型的蛋白质由2 871 个氨基酸组成),进而影响蛋白质的功能;多序列比对结果显示,FBN1 蛋白第2 471 位点脯氨酸在人、黑猩猩、恒河猴、鼠中高度保守,提示该变异可能具有致病性;在该家系中,该位点与临床表型共分离。在ClinVar 数据库中,有一例来源于马凡综合征患者的该位点的报道,对该位点的注释为致病变异。依据ACMG 指南,该位点为致病变异[致病变异分类非常强(PVS1)+中等证据2(PM2)+辅助证据1(PP1)]。
3 讨论
马凡综合征是一种常染色体显性遗传病,FBN1基因是唯一的致病基因[11]。马凡综合征的诊断标准一直在演进,1988 年柏林方案主要根据临床表现,到1996 年Ghent 方案,直至最新的2010 年修订版Ghent 方案,FBN1基因突变在疾病的诊断中越来越重要。修订版Ghent 方案提出,无家族史患者若主动脉根部直径Z 值≥2,并且合并晶状体脱位及FBN1基因突变,系统评分>7 等三项中的一项;或晶状体脱位合并FBN1基因突变并主动脉病变,即可临床诊断为马凡综合征[12]。本研究先证者体征包括身高过高、蜘蛛指、漏斗胸及关节活动过度;超声心动图提示主动脉根部增宽,A 型主动脉夹层,具有多项马凡综合征的临床表现,临床诊断为马凡综合征患者,家族中多位成员有胸主动脉夹层史并伴有马凡综合征体征,遂利用基因检测技术探索该家系的致病原因。
FBN1基因位于15q21.1,包含66 个外显子,编码2 871 个氨基酸。目前已经报道的与马凡综合征患者相关的FBN1突变已超过2 000 种[6]。FBN1基因无明显热点突变,据报道约12%的突变位点重复出现;编码区突变约占总突变的80%,非编码区突变约占总突变的20%。常见编码区突变有移码突变、错义突变和无义突变,移码突变占FBN1突变的18%左右[5]。近年来有多项研究在逐步勾勒马凡综合征基因型与表型的关系。Faivre 等[13]发现半胱氨酸错义突变与晶状体异位有关;Aoyama 等[11]在1995 年的一项研究中已经证明,导致原纤维蛋白-1蛋白单倍剂量不足的突变(HI-FBN1)与无事件生存期缩短和更严重的心血管并发症有关。此外,发生心血管事件的马凡综合征患者携带HI-FBN1突变频率更高[14]。Franken 等[15]证实,相比于DNFBN1(显性负效应)的患者,携带HI-FBN1突变的患者主动脉夹层和死亡的风险更高。2019 年,贡鸣等[16]对300 例中国马凡综合征患者的基因型与表型进行关联研究,发现主动脉夹层患者相较于主动脉根部瘤患者,携带移码突变频率更高(17% vs.4%,P=0.017)。随着越来越多基因型与临床表型之间的关系被揭示,利用基因检测技术在高危人群症状完全表现之前进行早期筛查,有助于疾病的早期诊断,对治疗方案制定有指导作用。
本研究通过对疑似马凡综合征的家系行基因检测,发现先证者及家系内存在相似临床表现者FBN1基因均存在相同移码突变(c.7412delC),该移码突变可能会造成编码蛋白的结构及功能改变,从而导致疾病发生。通过检索公共数据库,发现ClinVar 数据库中存在该变异位点的报道;dbSNP、ExAC、ESP6500、GnomAD 以及本团队内部数据库(800 例孤立性主动脉瘤患者全外显子测序数据库)均无该变异位点的频率报道;同时该变异存在家系共分离特点。依据ACMG 指南,该变异分级为致病变异。因此,根据该家系的临床表现及FBN1变异特点,并依据2010 年的新Ghent 诊断标准,先证者及其家系内携带该变异的成员均可确诊为马凡综合征;既往文献报道,携带移码变异的患者,更容易发生主动脉夹层,该先证者及家系内多位成年患者均发生主动脉夹层,提示该家系主动脉夹层高发,与以往研究报道一致。依据本研究的分子检测结果及相关家族史,可帮助临床医师对家系相关成员尤其是未成年患者进行随访,并合理选择治疗时机和方法。对携带变异的家系成员血压、血脂、血糖等心血管疾病危险因素的管理,有助于延缓急性事件的发生并改善预后[17]。样本的组织学检测对于疾病的病理特征分析有重要意义,本研究未获得相应的组织学样本并进行相关检测,有所缺陷,后续相关研究应重视组织学样本的收集。
本研究为该马凡综合征家系提供了分子诊断,为临床诊断和后续治疗选择提供了遗传学依据,进一步丰富了中国人群的马凡综合征遗传图谱。
利益冲突:所有作者均声明不存在利益冲突
