老年头颈部梭形细胞横纹肌肉瘤1例
2023-03-09莫小玲翁泽平胡云峰
莫小玲,翁泽平,胡云峰
1 临床资料
患者女,86岁,右侧面颊肿物半年。患者半年前右侧面颊无明显诱因出现一红色丘疹,无瘙痒及疼痛,未行诊治,逐渐增大。既往有胃溃疡出血、胆囊结石及胰腺炎病史多年,曾行胆囊切除术及胃大部切除术;否认高血压、糖尿病等慢性病史,家族史无特殊。查体:全身浅表淋巴结未触及,心肺腹未发现明显异常。皮肤科情况:右眼外下方约1 cm处可见约2.5 cm×2.5 cm大小的肿物,淡红色,圆形突出皮面,其表面溃疡、未见明显渗出,基底潮红,界清、质韧、无压痛(图1a)。予距肿物边缘0.3 cm处切除(图1b)并行病理检查。皮损组织病理示:部分表皮缺失,肿瘤位于真皮内,肿瘤细胞由大量的梭形细胞构成,呈编织状排列,浸润性生长,细胞异型,可见较多核分裂象(图2)。免疫组织化学示:p-CK(-),SMA弱阳性,S100(-),Desmin(+),MyoD1(+),Myogenin(-),CD34(-),ERG(-),Ki-67(约80%+)(图3)。诊断:梭形细胞横纹肌肉瘤。
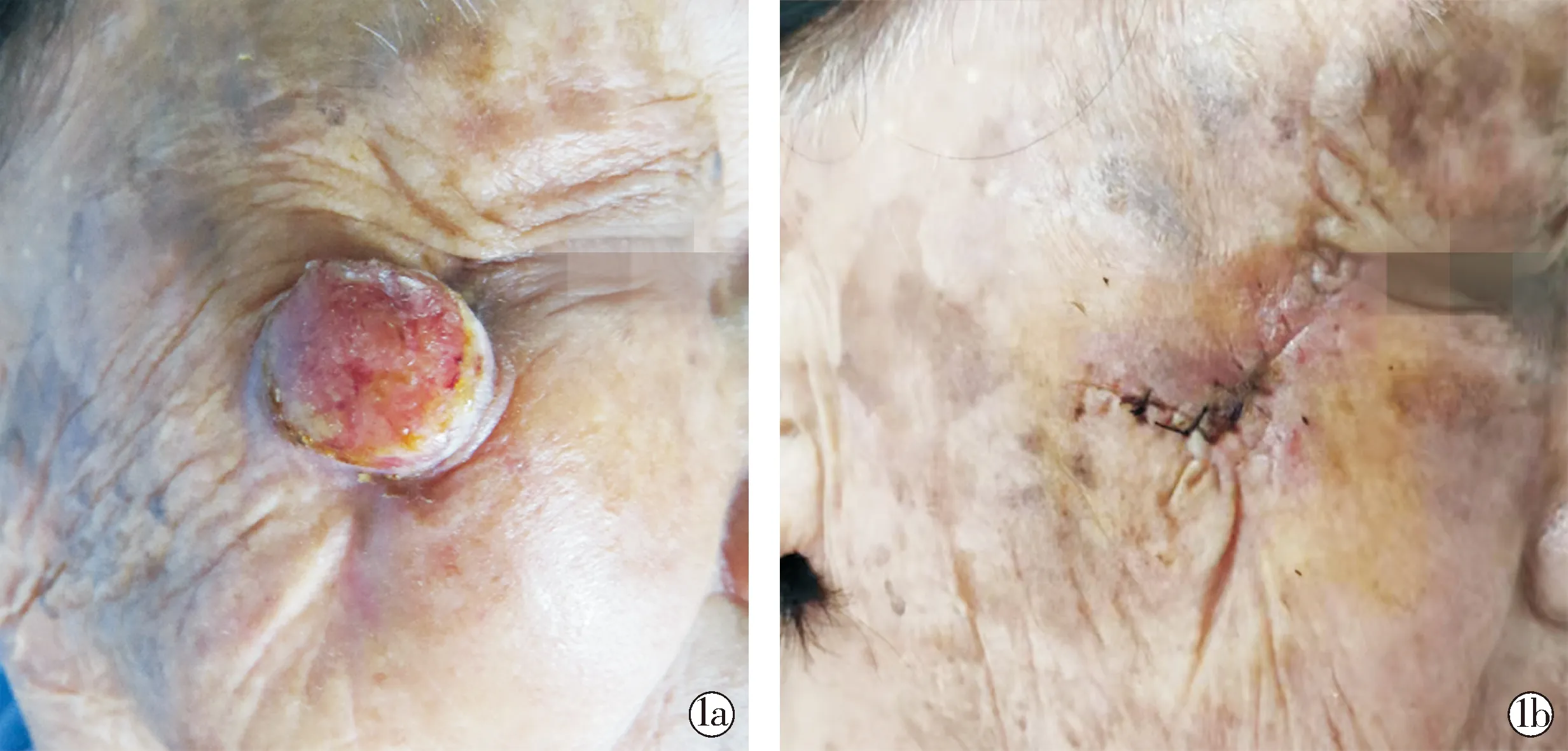
图1a 右面颊肿物;图1b 肿物切除术后Fig.1a The lump on the right cheek; Fig. 1b The mass after resection
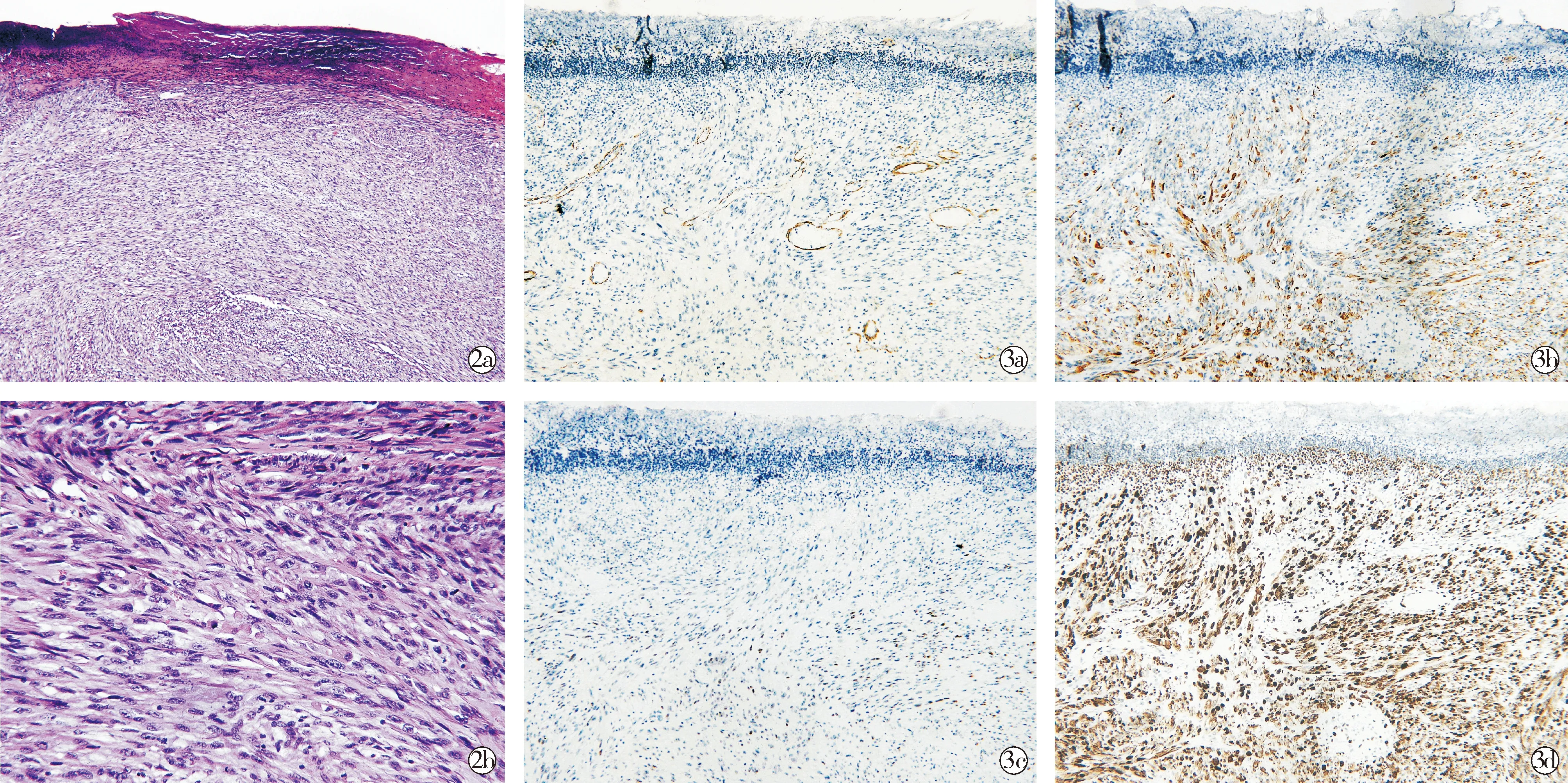
HE×40; HE×200;SMA(weakly positive); Desmin(+);MyoD1(+);Ki-67(approximately 80%+)
患者拒绝行基因检测、CT及彩超等检查。术后1个月切口愈合尚可,随访至今未见复发。
2 讨论
横纹肌肉瘤(rhabdomyosarcoma, RMS)属于骨骼肌分化的原始间叶恶性肿瘤。该病多见于儿童,在成人软组织肉瘤中仅占 3%[1]。梭形细胞横纹肌肉瘤(spindle cell rhabdomyosarcoma,SRMS)于1992年由Cavazzana等[2]首次描述为胚胎型的一种罕见变异。WHO软组织肿瘤分类(第4版)将RMS分为4型:胚胎性、腺泡性、梭形细胞/硬化性和多型性横纹肌肉瘤[3]。SRMS是RMS中较罕见的一个亚型,占所有RMS病例的5%~10%[4]。主要由束状排列的梭形细胞构成,儿童睾丸旁区多见,成人好发于头颈部[3]。病因及发病机制未完全明确。可能与基因变异相关,已发现的变异有:MyoD1基因突变,PIKC3A基因突变,VGLL2/NCOA2/CITED2重排,EWSR1/FUS-TFCP2、TPM3-NTRK1、SYPL1-BRAF、TOP2B-RAF1、EP300-VGLL3、MEIS1-NCOA2及HMGA2-NEGR1融合基因等[4-5]。其中MyoD1突变多见于较大的儿童和成人,是肿瘤侵袭性的标志。NCOA2和VGLL2重排多见于婴儿[4]。EWSR1/FUS-TFCP2融合是否为梭形细胞/硬化性横纹肌肉瘤的异常侵袭变体仍未明确[6]。
本例患者表现为右眼外下方肿物;组织病理主要由编织状排列的梭形细胞构成;免疫组织化学Desmin及MyoD1阳性,Ki-67约80%,其p-CK、S-100、CD34、ERG均阴性,可排外上皮、神经及血管来源的肿瘤,文献报告中有出现Myogenin阴性及SMA阳性的患者[7]。因此诊断为梭形细胞横纹肌肉瘤。
SRMS临床表现无特殊,通常为迅速生长的肿物。正确诊断需组织病理及免疫组织化学检查,必要时需行(分子遗传学)基因检测。组织病理见肿瘤主要由束状长梭形细胞构成,细胞核长、卵圆形、两端稍钝,胞质深嗜酸性,多数胞质内可见横纹,瘤细胞间可含胶原,可排列呈交织状等;部分病例可与硬化性、腺泡状等RMS合并存在。免疫组织化学可表达Desmin、MyoD1、Myogenin、Vimentin等。偶有异常表达WT1、CD99、CK、S-100、CD20、CD34及CD117等。本病常需与以下疾病鉴别:①梭形细胞鳞状细胞癌,组织病理与SRMS相似,但可累及表皮,肿瘤细胞间血管明显扩张;免疫组织化学可表达上皮、鳞状及肌源性标记物等;表达肌源性标记物时可行分子检测鉴别。②促结缔组织增生性黑素瘤,形态学上梭形细胞可呈束状排列,多与表皮关系密切;S-100、HMB-45及Melan-A阳性可鉴别。③平滑肌肉瘤,多发于成人腹膜后、四肢及躯干;形态学与本病相似,但无横纹肌母细胞,Masson染色胞质呈红色;且免疫组织化学不表达MyoD1和Myogenin。④恶性蝾螈瘤,多起源于神经纤维瘤病患者或周围神经;肿瘤内可具有恶性周围神经鞘瘤成分及横纹肌肉瘤成分,以前者为主;两种肿瘤成分分别表达神经源性及横纹肌的标志物。⑤成人型纤维肉瘤,好发于成人下肢, 其次是上肢和躯干;形态学上与SRMS相似,但间质胶原较多,Masson染色胶原纤维呈蓝色;免疫病理可见Vimentin阳性,但Desmin、MyoD1、Myogenin等均阴性[7-8]。
SRMS进展快,可转移。常用治疗方法为手术切除联合化疗或放疗,但目前无标准的化疗方案[1],多采用VAC方案化疗:长春新碱+放线菌素D+环磷酰胺。研究发现,VGLL2、TEAD1等基因融合者较少转移,随访存活良好。基因检测阴性者预后良好。MyoD1-突变型采用多模式治疗后复发、转移及死亡率仍较高;其预后与年龄和表型无关,儿童和成人预后都不理想[9]。本例肿物靠近右眼,因此予距肿物边缘0.3 cm处切除;患者拒绝行基因检测、CT及彩超等检查,遂未予化疗。随访至今未见复发。
头颈部SRMS较罕见,临床表现无特殊,组织病理与较多疾病相似,易误诊,免疫组织化学及基因检测可鉴别。基因突变类型与预后关系密切,早发现、早诊断是改善预后的关键所在。通过本例资料和文献复习,笔者希望提高对头颈部皮肤少见肿瘤的认识,以期为后来者诊断类似病例提供帮助。
