异唑类杀虫剂氟唑酰胺合成工艺研究
2023-03-06祝青波刘瑞宾刘安昌
李 宁,祝青波,刘瑞宾,刘安昌
(1.山东康乔生物技术有限公司 山东 博兴 256500;2.武汉工程大学 化工与制药学院,武汉 430074)
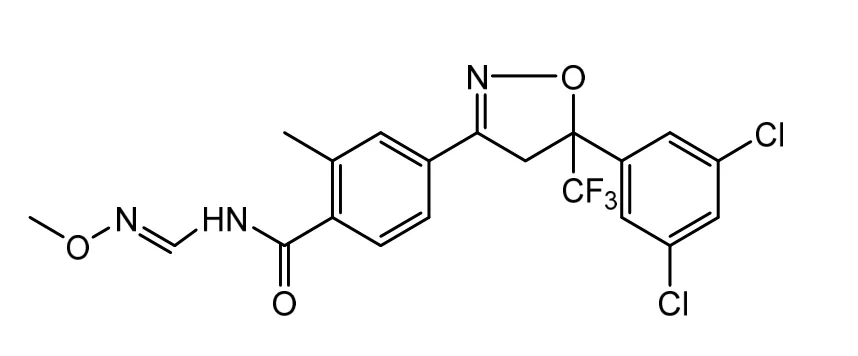
图1 氟唑酰胺的结构式
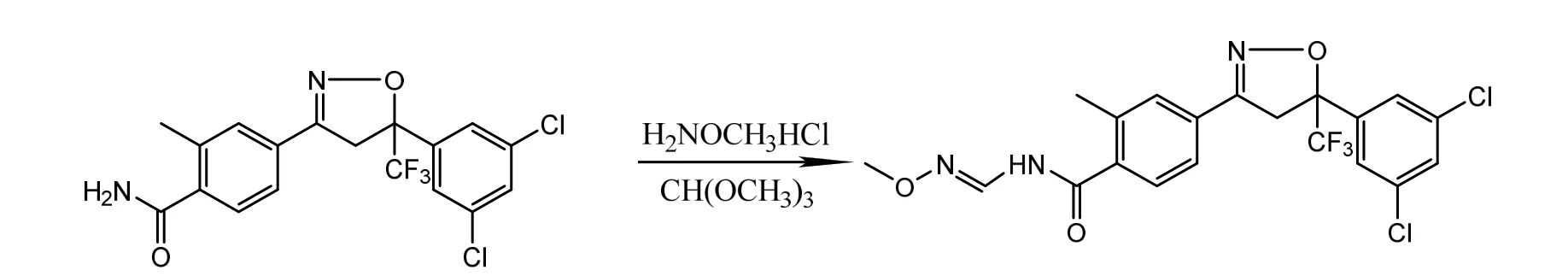
图2 氟唑酰胺的合成路线
上述关键中间体主要有2 条合成路线。
路线一:以2-甲基-4-乙酰基苯甲酸为原料,经氯化、胺化得2-甲基-4-乙酰基苯甲酰胺;然后在三乙胺的作用下与3′,5′-二氯-2,2,2-三氟苯乙酮缩合,盐酸羟胺成环得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰胺[4](图3)。
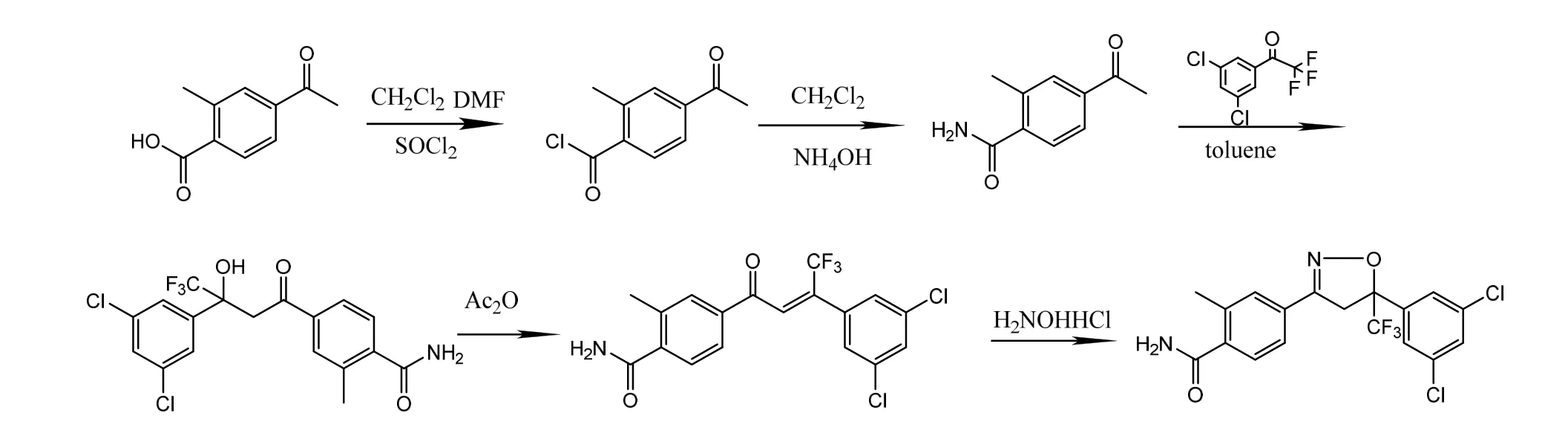
图3 4-(5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基)-2-甲基苯甲酰胺合成路线一
路线二:以2-甲基-4-乙酰基苯甲酸在月桂酸钠与3′,5′-二氯-2,2,2-三氟苯乙酮缩合,再与盐酸羟胺环化得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酸;最后经二氯亚砜酰氯化后与氨水反应得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰胺[5](图4)。

图4 4-(5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基)-2-甲基苯甲酰胺合成路线二
中间体3′,5′-二氯-2,2,2-三氟苯乙酮也有2 条合成路线。
路线一:以3,5-二氯苯甲酸为原料,在浓硫酸催化下酯化,得到3,5-二氯苯甲酸甲酯,然后与三甲基三氟甲基硅,在氟化铯作用下得到1-(3,5-二氯苯基)-2,2,2-三氟-1-三甲基甲硅烷氧基-1-甲氧基乙烷;然后在四丁基氟化铵作用下得到目的产物[6](图5)。该路线使用了昂贵的三甲基三氟甲基硅和氟化铯,生产成本高,不宜工业化。

图5 3′,5′-二氯-2,2,2-三氟苯乙酮合成路线一
路线二:以3,5-二氯溴苯为原料,经格氏化,与三氟乙酸甲酯反应共2 步得到目的产物(图6)。该路线工艺简单,原料易得[7]。

图6 3′,5′-二氯-2,2,2-三氟苯乙酮合成路线二
1 实验部分
1.1 主要试剂
2-甲基-4-乙酰基苯甲酸(实验室自制);3,5-二氯溴苯[前衍化学科技(武汉)有限公司];三氟乙酸甲酯(上海升得科技有限公司);羟胺50%水溶液(罗恩试剂);甲氧基胺盐酸盐、四丁基溴化铵(98%)(武汉格奥化学技术有限公司);其他试剂均为国药分析纯。
1.2 3',5'-二氯-2,2,2-三氟苯乙酮的合成
在装有搅拌器和温度计的500 mL 反应瓶中,加入150 mL 干燥的四氢呋喃(THF)和50.40 g(0.21 mol)金属镁,加热至40~50 ℃,滴加45.20 g(0.20 mol)的3,5-二氯溴苯,待反应完全后,冷却至室温,滴加56.80 g(0.40 mol)三氟乙酸甲酯,保温反应5~6 h。反应完全后,蒸出部分THF,用二氯甲烷(DCM)萃取2×50 mL,合并有机层,无水硫酸钠干燥,浓缩得黑色液体。减压蒸馏,收集68~83 ℃/2 mmHg 的馏分,得34.17 g 淡黄色液体,收率70.3%。
1.3 4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁酰基)-2-甲基苯甲酸的合成
在装有搅拌器和温度计的500 mL 反应瓶中,加入180 mL 水,17.80 g(0.10 mol)2-甲基-4-乙酰基苯甲酸,26.70 g(0.11 mol)3′,5′-二氯-2,2,2-三氟苯乙酮和1.20 g 月桂酸钠,加热至85~90 ℃,反应3~5 h。冷却,加入200 mL 乙酸乙酯萃取,用乙酸调pH=1~2,分出有机层,水层用3×50 mL 乙酸乙酯萃取,合并有机相,用3×50 mL 水洗,浓缩得黄色固体30.00 g,收率71%。
1.4 4-(5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基)-2-甲基苯甲酸的合成
将10.14 g(0.019 mol)4-[3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁酰基]-2-甲基苯甲酸三乙胺盐,0.35 g(0.003 mol)4,4-二甲氨基吡啶,60 mL 甲苯加入250 mL的反应瓶中,于50 ℃下滴加5.00 g(0.049 mol)乙酸酐搅拌10 h。冷却,加入50 mL 甲苯,分出有机层,加入50%盐酸羟胺2.62 g(0.02 mol),反应5 h。用2×60 mL 水洗,浓缩得稠状固体,用正己烷重结晶,得黄色固体5.20 g,收率65%。
1.5 4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰氯的合成
将20.90 g(0.05 mol)4-(5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基)-2-甲基苯甲酸溶液100 mL 甲苯溶液中,加入2 滴N,N-二甲基甲酰胺,升温至80 ℃,滴加11.90 g(0.20 mol)氯化亚砜。滴加完毕,在相同温度下继续搅拌1.5 h。反应完成后,减压蒸馏除去部分溶剂,然后加入50 mL 正己烷,在60 ℃搅拌30 min,冷却至室温结晶,过滤得白色结晶21.30 g。收率98%,熔点:140~143 ℃。1H NMR(CDCl3, 300 MHz) δ: 8.22 (d, J=8.7 Hz, 1H), 7.60 (d,J=8.7 Hz, 1H), 7.60 (s, 1H), 7.50 (s, 2H), 7.41(s,1H),4.10(d,J=17.4 Hz,1H),3.71(d,J=17.4 Hz,1H),2.64(s,3H).
1.6 4-(5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基)-2-甲基苯甲酰胺的合成
在装有搅拌器和温度计的四口反应瓶中,加入150 mL 四氢呋喃和21.80 g(0.05 mol)4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰氯,冷却至10~15 ℃,滴加12.50 g(0.10 mol)28%的氨水,滴加毕,在室温下继续搅拌18 h。反应完全后,减压蒸馏除去溶剂,残余物用150 mL乙酸乙酯萃取,分别用50 mL 水和饱和氯化钠水溶液洗涤、无水硫酸钠脱水干燥,减压蒸馏除去溶剂,得到19.70 g 橙色结晶的目标产物,收率95%。熔点:162~164 ℃。1H NMR(CDCl3,400 MHz)δ:7.45-7.55(m,6H),6.42(bs,1H),6.05(bs,1 H),4.12(d,J=17.0 Hz),1 H),3.70(d,J=17.0 Hz,1 H),2.51(s,3H)。
在装有搅拌器和温度计的四口反应瓶中,加入20.80 g(0.05 mol)4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰胺、53.00 g(0.35 mol)原甲酸三乙酯、6.25 g(0.075 mol)甲氧基胺盐酸盐和甲苯溶液200 mL,在50 ℃下搅拌24 h。反应完成后,向反应溶液中加入100 mL 甲苯。将反应溶液加热至60~65 ℃,用50 mL×3 水洗涤。减压蒸除去部分甲苯。冷却结晶。过滤,产品用少量甲苯洗涤,晶体真空干燥,得到19.20 g 白色晶体,收率81%。熔点:172~174 ℃。1H NMR(CDCl3,400 MHz)δ: 8.49(d, J=9.4 Hz, 1H), 7.77(d,J=9.2 Hz, 1H),7.61-7.48 (m, 5H), 7.43(t, J=1.8 Hz, 1H), 4.08 (d,J=17.2 Hz,1H),3.90(s,3H),3.70(d,J=17.3 Hz,1H),2.53(s,3H)。
2 结果与讨论
2.1 反应温度对3',5'-二氯-2,2,2-三氟苯乙酮收率的影响
在制备3′,5′-二氯-2,2,2-三氟苯乙酮的过程中,反应温度对3,5-二氯苯基溴化镁与三氟乙酸甲酯的反应效果至关重要。反应温度过高,格氏试剂反应太快,副反应增加,产品收率较低,反应温度较低时,反应速度有较慢,格氏试剂容易吸水,影响反应效果。通过实验条件优化,在室温25 ℃左右,反应效果最好,收率可以达到70%以上。
2.2 原甲酸三乙酯用量对氟唑酰胺收率的影响
2.3 氟唑酰胺核磁共振氢谱表征
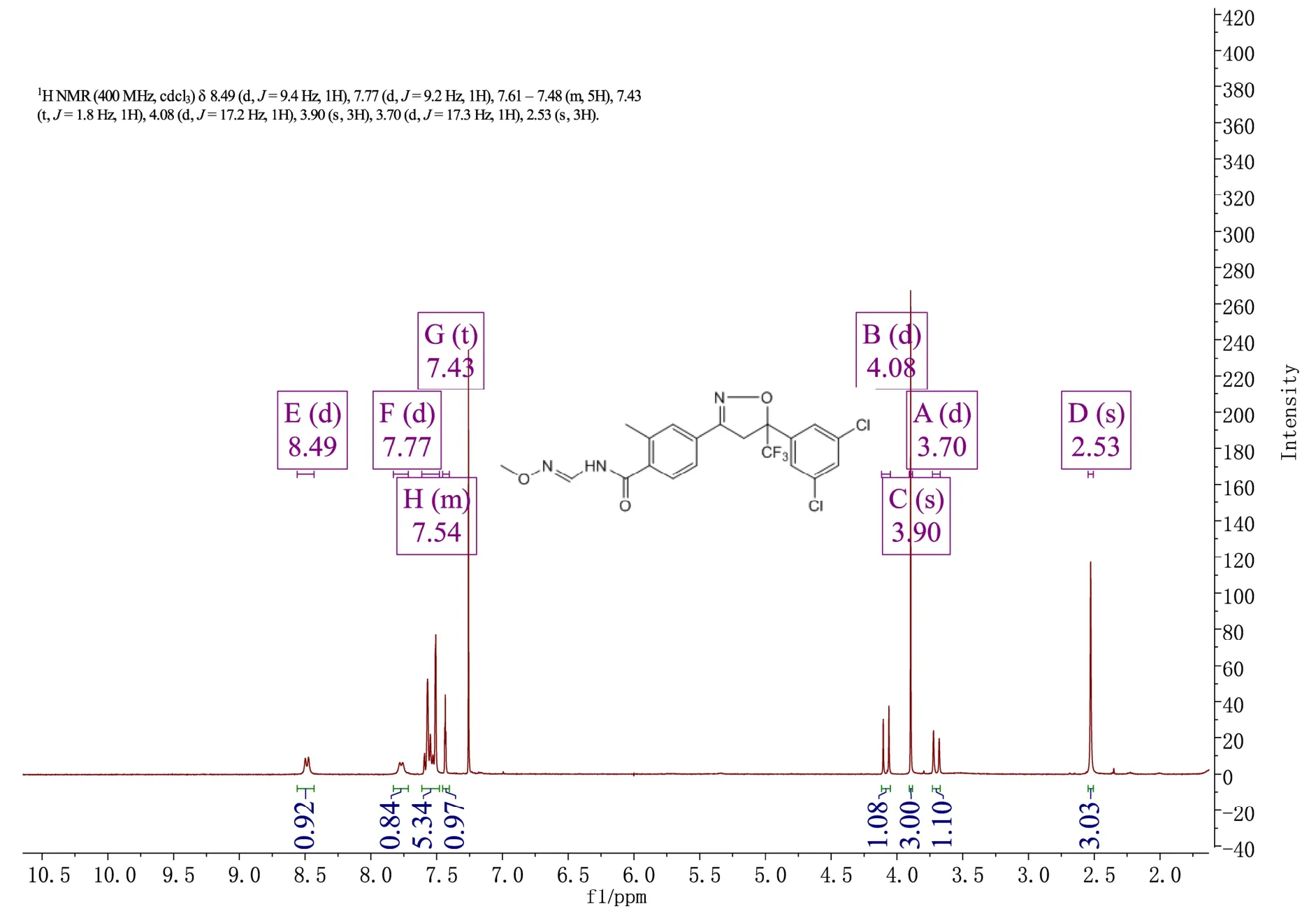
图7 氟唑酰胺核磁共振氢谱
3 结 论
⑴以3,5-二氯溴苯为原料,经格氏化,与三氟乙酸甲酯反应共2 步合成得到3′,5′-二氯-2,2,2-三氟苯乙酮;在月桂酸钠的催化下,2-甲基-4-乙酰基苯甲酸与3′,5′-二氯-2,2,2-三氟苯乙酮缩合,脱水,再与盐酸羟胺环化得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酸。
⑵4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酸与氯化亚砜酰氯化后,再和氨水反应得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异唑-3-基]-2-甲基苯甲酰胺;最后与原甲酸三甲酯,甲氧基胺盐酸盐反应得到目的产物氟唑酰胺。该路线工艺简单,反应条件温和,具有工业化应用价值。
