7-硝基靛红的高效液相色谱分析
2023-03-06肖瑛子黄超群庞怀林
肖瑛子,吴 若,唐 亮,黄超群,庞怀林
(1.江西天宇化工有限公司长沙分公司,长沙 410209;2.长沙嘉桥生物科技有限公司,长沙 410209;3.南通泰禾化工股份有限公司,江苏 南通 226407)
7-硝基靛红是由南通泰禾化工股份有限公司自主研发的环丙氟虫胺的重要中间体[1-3],是近期发展较快的重要精细化工中间体,其分子式为C8H4N2O4,化学结构式见图1。

图1 7-硝基靛红的结构式
目前关于7-硝基靛红的分析方法少。本文采用高效液相色谱法,将其与环丙氟虫胺工艺生产过程的原料和杂质进行分离,根据分离度与峰型对分析条件进行研究[4-5],选择了乙腈+纯水为流动相,建立了适用于7-硝基靛红样品的定量分析方法,具有分离效果好、操作简单快速等特点。
1 材料与方法
1.1 主要仪器与试剂
LC-20AT 型高效液相色谱仪配SPD-M20A 二极管阵列检测器和色谱工作站(日本岛津公司);ATY224 型电子天平(日本岛津公司);SXC-10JZ 型超声波清洗仪(湖南尚辛创仪器设备有限公司);UPH-1-10L 型纯水超纯水一体机(四川宜家水处理科技有限公司);0.22µm 滤膜(水系、有机系,彼西络科技有限公司)。
乙腈(色谱纯,韩国SK chemicals);7-硝基靛红标样,已知质量分数99.4%(长沙嘉桥生物科技有限公司,上海晓明检测技术服务有限公司定值);7-硝基靛红试样样品(江西天宇化工有限公司)。
1.2 试验方法
1.2.1 色谱条件
色谱柱为岛津Shim-pack GIST C18不锈钢柱(柱长250 mm,内径4.6 mm,内装5µm填充物);流动相为乙腈+纯水(15+85,体积比),经0.22 μm滤膜过滤和脱气,流速为1.0 mL/min;检测波长为270 nm;柱温为(40±2) ℃;进样体积为5µL。在该色谱操作条件下,7-硝基靛红的保留时间约为9.9 min。7-硝基靛红标样和试样高效液相色谱图见图2。

图2 7-硝基靛红标样和试样高效液相色谱图(500 μg/mL)
1.2.2 测定步骤
1.2.2.1 标样溶液的配制
称取0.05 g(精确至0.000 1 g)7-硝基靛红标样置于100 mL 容量瓶中,加入乙腈溶解,放入超声波清洗仪中超声3 min,待溶液冷却至室温后用乙腈定容,摇匀。
1.2.2.2 试样溶液的配制
称取含0.05 g(精确至0.000 1 g)7-硝基靛红试样置于100 mL 容量瓶中,加入乙腈溶解,放入超声波清洗仪中超声3 min,待溶液冷却至室温后用乙腈定容,摇匀。
1.2.2.3 测定
按上述操作条件,待仪器基线稳定后,连续注入数针标样溶液,待相邻2 针的峰面积相对变化小于1.0%时,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进样测定。
1.3 计算
将测得的2 针试样溶液以及试样前后2 针标样溶液中7-硝基靛红峰面积分别进行平均。试样中7-硝基靛红的质量分数ω(%)按式(1)计算:
式中:ω为试样中7-硝基靛红的质量分数(%);A2为试样溶液中7-硝基靛红峰面积的平均值;m1为标样的质量(g);P 为标样中7-硝基靛红的质量分数(%);A1为标样溶液中7-硝基靛红峰面积的平均值;m2为试样的质量(g)。
2 结果和分析
2.1 色谱条件的选择
为了实现7-硝基靛红与杂质的有效分离,根据其化学结构特点,对乙腈和纯水、甲醇和纯水、乙腈与磷酸水溶液、甲醇与磷酸水溶液4 种组合,在Shim-pack GIST C18色谱柱上进行比较选择,经过试验比较,确定流动相为乙腈与纯水的混合溶液,比例为15:85(体积比),流速为1.0 mL/min。在上述确定的流动相条件下,对Shim-pack GIST、ODS-3、Waters 3 款C18色谱柱进行比较选择,从峰型对称和分离效果来看,选择Shim-pack GIST C18色谱柱。
通过高效液相色谱仪的SPD-M20A 二极管阵列检测器进行紫外光谱扫描,得到7-硝基靛红的紫外吸收光谱图,其最大吸收波长为201 nm,次吸收波长为270 nm。通过多次试验,波长为270 nm 时7-硝基靛红及其他杂质峰都有很好的吸收且峰型平缓,因此本试验选择270 nm 为检测波长。
经过不同柱温的进样试验,柱温在30~40 ℃时7-硝基靛红保留时间的变化不显著;考虑到柱效以及高效液相色谱试样检测池池温,选择40 ℃作为此方法的分析温度。
在此条件下,基线平稳,7-硝基靛红与其他杂质得到较好地分离,且峰型对称。
2.2 分析方法的特异性
采用峰纯度来鉴别7-硝基靛红。7-硝基靛红标样和试样的最小峰纯度相似度均为1.00,最小峰纯度阈值处在0.99~1.00 范围内,分别为0.999 9 和0.999 4,最小峰纯度指数分别为0 和551,非负值,说明标样和试样色谱峰中均不含有杂质峰。
2.3 分析方法的线性相关性
分别称取6 份7-硝基靛红标样于6 个100 mL容量瓶中,加入乙腈溶解,放入超声波清洗仪中超声3 min,待溶液冷却至室温后用乙腈定容,摇匀。
以7-硝基靛红的质量浓度为横坐标,其峰面积为纵坐标绘制标准曲线,线性回归方程为y=18 459x-13 760,相关系数R2=1。结果见表1 和图3。由图可见,7-硝基靛红的质量浓度为51.89~233.19 μg/mL时有良好的线性关系。
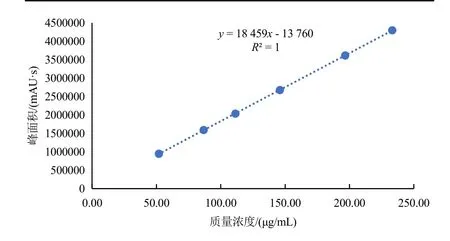
图3 7-硝基靛红的质量浓度与其相应峰面积的线性关系图
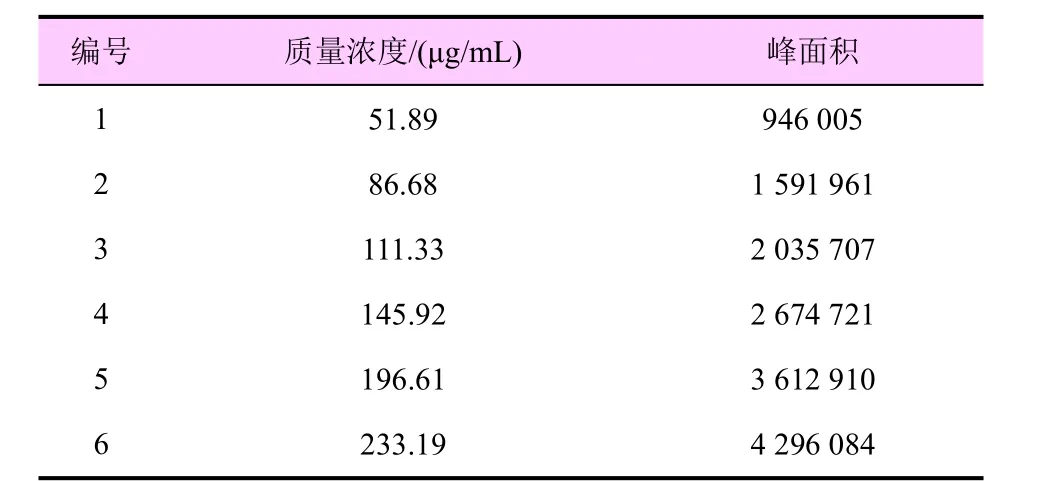
表1 7-硝基靛红的质量浓度及其相应峰面积值
2.4 分析方法的精密度
称取已知含量试样6 个,按上述色谱操作条件进行分析,测得7-硝基靛红的质量分数,试验结果详见表2。

表2 7-硝基靛红试样的精密度试验结果
7-硝基靛红的标准偏差为0.16%,变异系数为0.18%,说明方法精密度良好,能满足日常的定量分析。
2.5 分析方法的准确度
分别称取0.025 g(精确至0.000 1 g)7-硝基靛红试样置于6 个100 mL 容量瓶中,加入不同质量的7-硝基靛红标样(精确至0.000 1 g),按1.2 节中溶液的配制方法进行配制后进行测定,结果见表3。由表3 回收率的测定可知,7-硝基靛红回收率为100.12%~100.86%,平均回收率为100.60%,均在98%~102%之间,说明该方法准确度良好,能满足日常的定量分析。

表3 7-硝基靛红的准确度试验结果
2.6 非分析物的干扰
以不含有效成分的空白溶剂作为空白试样的高效液相色谱图如图4,通过其色谱图与图2 对比确认,空白试样中不含有效成分的干扰物质,能够确保防止有效成分中其他物质的干扰,避免分析方法出现误差。

图4 7-硝基靛红溶剂空白的高效液相色谱图
3 结 论
本文建立了高效液相色谱法测定7-硝基靛红的定性定量方法。经试验验证,该方法线性范围宽、重现性好、准确度高,可以快速地对7-硝基靛红进行定性和定量分析,能满足对产品质量控制的要求。
