WNK-SPAK/OSR1-CCCs信号通路与缺血性脑卒中关系的研究进展
2023-03-02张建云李婧雯梅紧紧王艺莹段昭远苏凯奇综述李瑞青审校
张建云,李婧雯,梅紧紧,王艺莹,段昭远,高 静,苏凯奇 综述 郭 健,李瑞青, 审校
脑卒中是由急性脑血管损伤引起的局部脑功能障碍[1],临床以高发病率、高致残率、高死亡率为主要特征[2]。据统计,我国脑卒中现患人数高居世界首位,已成为导致成年居民死亡和残疾的首位病因[3]。目前对于缺血性脑卒中的研究多以宏观角度为主,具体发病机制尚不十分明确。有研究表明,丝氨酸/苏氨酸蛋白激酶(with-no-lysine kinases, WNK)-Ste20相关的富含脯氨酸-丙氨酸激酶(Ste20-related proline-alanine-rich kinase, SPAK)/ SPAK同源物氧化应激反应激酶1(oxidative stress-responsive kinase 1,OSR1)-阳离子-氯离子共转运体(cation-chloride cotransporters, CCCs)信号通路与缺血性脑卒中联系密切[4]。而抑制NKCC1的表达,增强KCC2和KCC3的表达,维持细胞内较低的Cl-浓度,是CCCs正常表达的关键[5-6]。现通过梳理WNK-SPAK/OSR1-CCCs信号通路在神经细胞中的表达来探讨其对缺血性脑卒中的作用机制,为其临床治疗缺血性脑卒中的研究提供新的思路和药物靶点。
1 NKCC1结构功能及其与缺血性脑卒中的关系
1.1 NKCC1结构与功能NKCC1属于SLC12A家族,由SLC12A2基因编码[7],该基因位于第5号染色体[8]。人类SLC12A2基因至少编码三种剪接变体,编号分别为NM 001046(SLC12A2 v1, NKCC1a)、NM 001256461(SLC12A2 v2, NKCC1b)和NR 046207(SLC12A3 v3)[9]。NKCC1a和NKCC1b转录本编码蛋白质130 ku,具有预测的12个跨膜结构域和两个大的细胞内N端和C端[10]。资料显示,糖基化分布于NKCC1的N端,其中,复杂型N-糖基化约含有25%,核心/高甘露糖型和混合型N-糖基化约含有75%[11]。此外,NKCC1翻译后约10%到达质膜,以核心/高甘露糖型N-糖基化为主,其余90%的NKCC1存在于细胞中,复杂型N-糖基化是转运体发挥作用的必要条件。但NKCC1的N-聚糖性质和复杂N-聚糖对其质膜插入的影响尚不清楚,还有待进一步研究。此外,NKCC1主要在外周神经系统、背根神经节以及三叉神经节表达,在中枢神经系统也有大量表达,但不同的发育时期NKCC1的表达不同,NKCC1在哺乳动物发育早期表达相对较高,并且伴随发育不断成熟而逐渐降低[12]。其在正常情况下通过继发性主动转运的形式介导Na+、K+、Cl-内流,可以调节抑制性突触传递、维持细胞体积[13]。
1.2 NKCC1与缺血性脑卒中的关系NKCC1广泛存在于大脑的神经细胞中,在调节神经细胞功能方面起着关键作用,主要参与Cl-平衡的维持和神经元的兴奋功能[14],并通过GABA能信号发挥调节和修复神经损伤的功能[15]。然而,缺血性脑卒中后NKCC1的表达将会发生改变[16]。研究[17-18]表明,缺血性脑卒中后,神经元中NKCC1的表达增加,这种表型类似于不成熟的神经元。Mu et al[19]通过建立内皮素-1(endothelin 1,ET-1)脑卒中模型来诱导局灶性缺血性脑卒中的大鼠,发现在皮层周围其NKCC1的活性明显增加,而NKCC1活性增加可进一步诱导Na+内流,使神经细胞内渗透压升高,脑组织摄取水分增多,从而引发脑水肿。此外,缺血性脑卒中后,NKCC1 mRNA基因表达增加,NKCC1蛋白的激活在缺血性脑卒中病程中也起重要作用。Chen et al[20]通过建立小鼠短暂性大脑中动脉闭塞模型(MCAO)发现,NKCC1或其上游调节器WNK3的基因缺失后,NKCC1活性下降,Na+内流减少,神经细胞内Cl-浓度降低,渗透压降低,脑组织摄取水分减少,可使脑梗死、脑水肿和白质损伤的程度明显降低。同理,通过抑制NKCC1活性可减少细胞内Na+浓度并减轻水肿,恢复神经元内Cl-平衡。Xu et al[21]通过动物实验发现,与健康对照组相比,在大鼠局灶性脑缺血后给予NKCC1抑制剂布美他尼(Bumetanide)治疗,其神经细胞内Na+内流减少,渗透压降低,这是因为布美他尼通过与NKCC1跨膜区的结合点对接发挥作用,可抑制NKCC1的活性,刺激KCC3的表达,从而减少神经元中的Na+和Cl-浓度,增加神经细胞的存活,减轻脑水肿,促进缺血性脑卒中后神经发生和行为恢复。由此可见,NKCC1表达的增加可引起细胞内Cl-稳态失衡,不利于脑功能的恢复,而加入影响其表达的抑制剂可起到改善脑功能的效果。
2 KCC2结构功能及其与缺血性脑卒中的关系
2.1 KCC2结构与功能KCC2是一种约140 ku的糖蛋白,是SLC12A家族中的一员,由24个外显子的SLC12A5基因编码,该基因分配给人类染色体带20q12→q13.1,以及小鼠染色体带波段5G2→G3[22]。有研究[23]显示,KCC2基因编码有两个亚型,并分别命名为KCC2a和KCC2b,两者在氨基酸末端(N端)的结构不同。这两种KCC2异构体在新生儿大脑中有类似的表达模式,但KCC2b在成年大脑中成为主要的异构体[24-25],主要参与γ-氨基丁酸(γ-aminobutyric acid, GABA)能神经元由去极化状态转变为超极化状态这一过程,维持氯离子稳态,调节GABA能神经元的发育。此外,KCC2蛋白具有12个跨膜段的膜拓扑结构,两侧是亲水的N端和C端结构域,在跨膜段5和6之间有一个大的糖基化胞外环[22]。KCC2有一个15个残基的C端等渗结构域,被称为ISO结构域,该结构域是等渗条件下的钾氯共转运的必要条件[26]。但尚未有证据证明KCC2表达的分子机制受何种因素调控,这些因素包括同家族的其他成员调节、自身调节、受到神经递质的调节等,仍需进一步探讨。
2.2 KCC2与缺血性脑卒中的关系KCC2是成熟神经元中Cl-外流的主要转运体,其功能和表达受神经元活动的调节[27]。在某些情况下,神经元的激活可能会上调KCC2以加强突触抑制的平衡。然而,在缺血性脑卒中后痉挛发生时,KCC2的表达便会受到抑制,从而改变运动神经元兴奋和抑制的平衡,进而导致兴奋性毒性或异常活动[28]。GABA是中枢神经系统内重要的抑制性神经递质,在KCC2表达上调、NKCC1表达下调时,细胞内Cl-浓度降低,使GABA能神经元发挥抑制作用,可以有效防止痉挛的发生。而缺血性脑卒中后, KCC2表达下调、NKCC1表达上调,细胞内Cl-浓度增加,Cl-稳态失衡,GABA抑制作用减弱,从而引发痉挛,WNK-SPAK/OSR1在此过程中发挥重要的调控作用。Toda et al[29]发现,在局灶性脑损伤后,KCC2表达下调,引起细胞内Cl-浓度增加,导致Cl-稳态失衡,GABA抑制作用减弱,且KCC2也影响缺血性脑卒中后运动神经元质膜上的Ser940去磷酸化,这两种效应使运动神经元的兴奋性增强,从而导致卒中后痉挛的发生。此外,翻译后修饰机制也参与调节KCC2的功能[30-32]。Wu et al[33]发现,KCC2的调节功能可以通过改变共识位点的磷酸化状态来实现。具有激酶活性的WNK3通过增强KCC2的磷酸化而抑制其活性,从而增加细胞内Cl-浓度,促进对GABA能运动神经元的兴奋性反应。相反,无激酶活性的WNK3介导的KCC2的激活降低了细胞内Cl-浓度,促进了抑制性GABA信号的传递,使GABA能神经元发挥抑制作用,从而防止痉挛的发生。此外,通过药物干预增加KCC2活性,可恢复由于痉挛发生时减弱的GABA能运动神经元的突触后抑制作用,起到缓解痉挛的效果[34]。Ma et al[35]通过观察新生大鼠缺血缺氧的脑部发现,缺血性脑卒中早期应用地西泮治疗,可减轻神经细胞损伤,并有效改善长期的学习和记忆能力。这表明地西泮可以防止KCC2的下调,保持细胞内低氯状态,从而使GABA能运动神经元发挥抑制作用,进而改善神经功能。这也从侧面说明了KCC2的表达增强对神经功能的恢复以及痉挛的缓解起重要作用。具体机制见图1。
3 KCC3结构功能及其与缺血性脑卒中的关系
3.1 KCC3结构与功能KCC3是SLC12A家族的成员,由SLC12A6基因编码[36]。SLC12A6基因在5cM(5q13-15)区间内映射到人类染色体15q1.4[37]。SLC12A协同转运蛋白的细胞内末端包含重要的磷酸化位点,参与转运激活或失活。KCC3有两个主要的磷酸化位点,分别为T1048和T991,它们位于羧基末端[38]。研究[39]显示,KCC3与其他KCC家族高度同源,在氨基酸序列上有约73%的一致性,与Na+-耦合的NKCC1有中等程度的同源,与其他转运体有约29%的一致性。当在比对研究中排除N端时,两者的相同性分别增加到76%和32%。研究表明,KCC3在多种中枢神经系统(central nervous system, CNS)和周围神经系统(peripheral nervous system, PNS)细胞类型中表达[40]。在成年期,KCC3在CNS中广泛表达[41],以杏仁核和下丘脑中KCC3表达最高。在海马体中,KCC3位于中间神经元内[42]。对于成人PNS,KCC3在坐骨神经和背根神经节(dorsal root ganglion, DRG)中的表达量极低[43]。与NKCC1功能相反,KCC3通过继发性主动转运的形式介导Na+、K+、Cl-外流,二者相互拮抗,共同参与神经细胞抑制性突触传递以及细胞体积的调节。然而,KCC3转运所需的离子转运位点的数量仍未可知,需继续研究证实。
3.2 KCC3与缺血性脑卒中的关系在神经元功能调节中,KCC3的激活与NKCC1的抑制具有类似的生理作用。神经元中KCC3的表达需要依赖NKCC1的发挥,以完成对中枢神经系统的细胞平衡进行生理性调节[38, 44]。中枢神经系统中神经元抑制主要由GABA介导,而甘氨酸在较小程度上介导。GABA与Cl-可渗透的GABAA受体(GABAAR)结合后,受体离子通道打开,引起Cl-的流动[45]。Lucas et al[46]在一项小鼠模型中发现,同时抑制NKCC1表达和刺激KCC3表达,可促进成年小鼠感觉神经细胞内的Cl-浓度下降,维持神经细胞Cl-稳态,这表明它们参与GABA能/甘氨酸转换效能,引发GABA介导的超极化反应,从而介导抑制性GABA能神经传递效应,避免神经细胞过度兴奋,进而缓解痉挛的发生。此外,KCC3可能在调节细胞体积方面具有重要作用[39],其机制是在细胞肿胀的低渗条件下,细胞会激活调节性体积减少反应(regulatory volume decrease, RVD),此时,WNK-SPAK/OSR1激酶保持不活跃状态,引起NKCC1和KCC3去磷酸化,从而刺激KCC3表达和抑制KCC1表达,进而引起水和Na+、Cl-流出细胞,细胞体积减少。Byun et al[47]发现,刺激KCC3可参与神经系统的细胞体积调节,证明KCC3具有保护神经的作用,随后其研究进一步确定,通过敲除KCC3来抑制小鼠神经细胞中KCC3的表达,可引起细胞肿胀,最终导致神经变性。除上述在1.2项中所提到的布美他尼能够刺激KCC3表达外,Adragna et al[48]在细胞培养实验中发现,KCC3a蛋白的羧基末端用丙氨酸取代Thr 991和Thr 1048残基后,阻止了取代位点的抑制性磷酸化,从而引发KCC3a mRNA的表达增加,进而促进神经功能的恢复。以上研究表明,KCC3具有调节细胞体积和Cl-浓度的双重作用,为探讨其在缺血性脑卒中期间的发病机制提供了可靠证据。
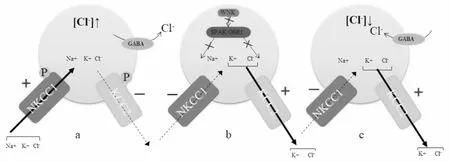
图1 WNK-SPAK/OSR1-CCCs信号通路在缓解痉挛中的作用
4 WNK-SPAK/OSR1-CCCs结构功能及其与缺血性脑卒中的关系
4.1 WNK-SPAK/OSR1-CCCs结构与功能WNK-SPAK/OSR1-CCCs信号通路与丝氨酸/苏氨酸蛋白激酶(with-no-lysine kinases, WNK)和下游Ste20相关的富含脯氨酸-丙氨酸激酶(ste20-related proline-alanine-rich kinase, SPAK)或SPAK同源物氧化应激反应激酶1(oxidative stress-responsive kinase 1,OSR1)以及阳离子-氯离子共转运体(cation-chloride cotransporters,CCCs)有关。WNK家族感知细胞内Cl-浓度、细胞外渗透压和细胞体积的变化,并将这些信息转导至CCCs,以维持细胞Cl-稳态[49]。研究[50]显示,缺血性脑卒中后,WNK通过SPAK和OSR1激酶途径,调节NKCC1、KCC2和KCC3的表达。WNK是一种Na+、K+耦合的Cl-导入器和K+耦合的Cl-导出器[51]。其异构体在细胞内低渗透压或Cl-水平低时磷酸化(激活),并随后使其下游的激酶SPAK和(或)OSR1磷酸化[52],激活的SPAK和(或)OSR1通过蛋白质磷酸化刺激NKCC1表达,并通过相互调节机制抑制KCC2、KCC3磷酸化[53]。在体积调节过程中,低渗环境引起细胞吸水膨胀,在正常条件下可触发调节性容积减少反应(regulatory volume decrease, RVD),WNK-SPAK/OSR1激酶保持不活跃状态,NKCC1和KCC3去磷酸化,导致KCC3激活和NKCC1抑制,K+、Cl-与水外流,细胞体积减少。相反,高渗环境引起细胞失水皱缩,触发调节性容积增加反应(regulatory volume increase, RVI),使WNK-SPAK/OSR1激酶活化,引起CCCs磷酸化,导致NKCC1激活和KCC3抑制,Na+、K+、Cl-通过NKCC1与水一起内流,从而恢复细胞体积。WNK-SPAK/OSR1激酶对CCCs的调节和两者的相互协调作用,由WNK和CCCs中的RFXV/I基序与SPAK和OSR1中保守的羧基末端对接结构域之间的相互作用所触发[4]。上述CCCs的反向调节由同一激酶-磷酸酶信号通路所驱动,可协调细胞内Cl-的外流和内流,从而维持细胞内Cl-的平衡[5],避免不必要的ATP消耗。
4.2 WNK-SPAK/OSR1-CCCs与缺血性脑卒中的关系WNK和SPAK/OSR1激酶在中枢神经系统中大量表达。缺血性脑卒中后,NKCC1、KCC2和KCC3作为WNK-SPAK/OSR1激酶的下游靶点,都通过WNK-SPAK/OSR1激酶磷酸化,从而导致NKCC1的激活与KCC2和KCC3的抑制[50, 54]。研究[55]表明,人类有四个WNK激酶(WNK1、WNK2、WNK3和WNK4),其中,WNK3在大脑中高度表达。Begum et al[56]通过建立小鼠MCAO模型,观察其脑内角质层神经细胞和初级少突胶质细胞发现,WNK3和SPAK激酶受到刺激,并进一步确定脑缺血促进了WNK3-SPAK/OSR1催化T环和NKCC1刺激位点(Thr203/Thr207/Thr212)的过度磷酸化,因此NKCC1在脑细胞中的表达增加。这表明WNK3-SPAK/OSR1-NKCC1信号通路在缺血性脑卒中期间发挥重要作用,阻断这一通路可减少NKCC1在大脑中的表达,预防缺血性脑卒中后的神经细胞死亡。此外,在对低Cl-浓度渗透压的反应中,WNK的异构体通过磷酸化被激活,激活的WNK异构体使相关的下游激酶SPAK或OSR1磷酸化,SPAK和(或)OSR1即被激活,激活的SPAK和(或)OSR1通过蛋白磷酸化刺激NKCC1,抑制KCC2和KCC3表达。Zhang et al[57]在一项实验性缺血性脑卒中研究发现,与WT小鼠相比,WNK3基因敲除和SPAK基因敲除小鼠在缺血性卒中后的梗死体积和脑水肿有效减少。考虑原因可能是WNK3和SPAK激酶通过增强NKCC1表达对缺血性脑卒中的恢复产生不利影响,而敲除它们便可增强KCC3表达,使水分伴随Na+和Cl-流出神经细胞,细胞体积减少,避免了神经细胞损伤和脑水肿,促进缺血性脑卒中后神经功能的恢复。同理,Josiah et al[58]采用支架杂交策略开发了一种非ATP竞争性SPAK抑制剂ZT-1a,发现ZT-1a可通过减少脑内SPAK依赖性磷酸化来抑制NKCC1和刺激KCC3,进而特异性抑制这一信号通路[59],改善缺血后CCCs的磷酸化,减轻脑水肿,进而改善神经功能。以上研究表明,WNK-SPAK-NKCC1基因敲除及其抑制剂对脑水肿和缺血性脑卒中的治疗具有潜力,可显著改善脑功能。见图2。

图2 WNK-SPAK/OSR1-CCCs信号通路与在细胞体积调节中的作用
5 总结与展望
阳离子-氯离子共转运体在减少细胞水肿、降低神经元内Cl-浓度和缓解痉挛方面发挥重要作用。CCCs的表达受其上游调节器WNK-SPAK/OSR1激酶的调节,与缺血性脑卒中的发生和发展有关。无论是在缺血性脑卒中发作期间还是在缺血性脑卒中后,NKCC1和KCC3的激活受WNK-SPAK/OSR1激酶的调控,导致NKCC1表达的增加和KCC3表达的抑制,这一过程在维持细胞体积方面发挥重要作用。而KCC2在维持神经细胞内Cl-平衡方面起着重要作用,缺血性脑卒中后KCC2表达的降低导致细胞内Cl-从低浓度到高浓度转变,神经细胞内Cl-稳态失衡,从而使GABA能运动神经元从抑制状态转为兴奋状态,进而导致痉挛的发生。因此,通过抑制WNK-SPAK/OSR1激酶磷酸化,进而抑制神经细胞内NKCC1的表达,提高KCC2和KCC3的表达是预防缺血性脑卒中后细胞水肿和痉挛的一个有效途径。本文探讨的CCCs抑制剂和激活剂对缺血性脑卒中的治疗具有潜在的疗效,故以阳离子-氯离子共转运体为治疗靶点可能为脑卒中的临床治疗提供治疗方向[60-61]。
