Engineering of Ag@Pd/Al2O3 with varied Pd-shell thickness: Dynamic evolution of ligand and strain effects on acetylene selective hydrogenation
2023-02-28MingboYangTianxingYangRuiMaShaLiYufeiHeDianqingLi
Mingbo Yang, Tianxing Yang, Rui Ma, Sha Li, Yufei He,4,5,, Dianqing Li,4,5
1 State Key Laboratory of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China
2 Department of Medicine and Chemical Engineering, Lianyungang Technical College, Lianyungang 222006, China
3 Chemistry and Chemical Engineering Guangdong Laboratory, Shantou 515031, China
4 Quzhou Institute for Innovation in Resource Chemical Engineering, Quzhou 324000, China 5 Beijing Engineering Center for Hierarchical Catalysts, Beijing University of Chemical Technology, Beijing 100029, China
Keywords:Catalyst Hydrogenation Nanostructure Shell-thickness-dependent performance Ligand and strain effect
ABSTRACT Bimetallic nanoparticles exhibit a synergistic effect that critically depends on their surface composition,but such promotion mechanisms become vague with varying surface compositions.Here, alumina supported Ag@Pd core-shell and PdAg alloy structure with controlled size and surface compositions were prepared to demonstrate synergetic mechanisms, particularly, ligand and strain effects on activity and ethylene selectivity for acetylene hydrogenation.The performance evaluation indicates that Ag@Pd catalysts with well-controlled Pd-shell thickness can effectively lower apparent activation energy and improve ethylene selectivity.Hydrogenation activity increases from 0.019 to 0.062 s-1 with decreasing Pd-shell thickness under mild conditions,which is 3-6 times higher than their alloyed and monometallic counterparts.Combined characterizations and density functional theory are conducted to reveal such shell-thickness-dependent performance.The ligand effect arising from Ag alloying in the interface of Ag@Pd2ML observes the strongest binding of acetylene,but it diminished sharply and the strain effect gets more prevailing with increasing shell thickness.The competition of ethylene desorption and deephydrogenation were also investigated to understand the selectivity governing factors,and the selectivity descriptor (0.5BE(C2H4) - BE(H)) was built to match the contribution of ligand and strain effect on the different surfaces of Pd-Ag bimetallic NPs.The exploration of synergetic mechanisms among bimetallic NPs with varied structure and surface compositions in this work can help us to deepen the understanding catalyst structure-activity relationship and provide a feasible way to optimize the overall catalytic performance.
1.Introduction
Ethylene is one of the most produced basic chemical building blocks.The commercial synthesis of ethylene is based on the cracking of oil fractions or light-saturated hydrocarbons, but the trace of acetylene is also produced, which is a pernicious ingredient for the downstream production of polyethylene.Thus, the elimination of trace acetylene in refined ethylene feedstock is an indispensable process for obtaining polymer-grade ethylene.Semi-hydrogenation of acetylene to ethylene on supported catalysts has been wildly applied, due to its economic efficiency.To this end,Pd-based materials are often used as hydrogen is easily dissociated on the Pd surface,and the selectivity of ethylene can be satisfied at low acetylene conversions[1-3].However,the reaction product rapidly shifts towards ethane near full conversion of acetylene.It is believed that acetylene hydrogenation on the Pd surface involves the sequential hydrogenation of acetylene and its subsequent hydrocarbon intermediates.Once ethylene is formed on the Pd surface, it can desorb or react further, which will lead to the formation of ethylene and by-product ethane, respectively.Besides, ethylene from the gas phase can also adsorb on the Pd surface and has the chance to react.Hence, a selective hydrogenation catalyst should have a higher activation barrier for the hydrogenation of ethylene than the barrier for ethylene desorption to restrain the deep-hydrogenation of ethylene.
Several factors may tune the preference between these two competitive steps (desorption and deep-hydrogenation of ethylene),resulting in differences in product distribution.In the acetylene hydrogenation reaction, it is recognized that the presence of large ensembles of Pd sites on the surface leads to a decrease of ethylene selectivity.Therefore, isolation or dilution of surface Pd atoms was proposed to weaken the adsorption of intermediate ethylene and increase ethylene selectivity, such as TiO2supported Pd1singe-atom,graphdiyne anchored Pd3trimer atoms,and other novel nanostructures of Pd sites [4-11].In the meanwhile, the modification of the electronic structure, in terms of ligand effect,can also regulate the adsorption strength between reactive species and active sites [12].Formation of bimetallic alloy by the addition of promoter metal,like Ag[13-16],Ga[17-19],Cu[20,21],In[22],Bi [23] and Zn [24-26], has been wildly studied to enhance ethylene selectivity.Taking alumina-supported Pd-Ag catalyst as an example,which is a preferential selection in this industrial process due to its satisfactory olefin selectivity compared to pure Pd catalyst [27].Tsapina et al.[28] studied the Pd-Ag bimetallic catalysts with XAS characterization and found that the distance between neighboring Pd atoms was stretched from 0.275 nm to the range of 0.282-0.299 nm, which prevents ethylene from adsorbing in a strong multisite mode and improves olefin selectivity.Also,electronic-rich Pd atoms inducing from Pd-Ag bonds promote the desorption of ethylene.However,the enhanced selectivity is generally achieved at the expense of hydrogenation activity, due to the increasing barrier for H2dissociation and weaker adsorption of reactants[29].The adjustment of hydrogenation activity and olefin selectivity arising from the addition of non-active metals is just like a see-saw relationship, which limits the optimization of the overall catalytic performance.
As a part of the local chemical environment,recent experiment and density functional theory (DFT) results suggest that the subsurface region plays a unique role in the hydrogenation reaction.For example, the subsurface C induced from reaction process can dominate desorption ability of ethylene and reaction path by modifying the energy of Pd 3d unfilled orbitals, leading to higher rates and less deep hydrogenation [30].While, the formation of PdHxphases under the reaction conditions are responsible for the change of activity, depending on the H/Pd ratios.Very recently,Petra E.de Jongh and co-workers[31]studied the synergistic effect in Au@Pd nanorod catalysts with thick Pd-shell and found that the turnover frequency (TOFs) of core-shell catalysts were up to 50 times higher than their alloy and monometallic counterparts in butadiene hydrogenation,while maintaining comparatively higher butene selectivity.Next, Lu’s work [32] suggests that the hydrogenation activity-selectivity trade-off in Pt nanoparticles (NPs)can be broken by fabrication of Au@Pt bimetallic catalyst with monolayer Pt shell.The high activity results from modification of platinum 5d-band center through Pt lattice expansion and ligand effect, and the high selectivity can be attributed to the exposure of more terrace sites on Pt surface shell.These results suggest that several subsurface layers in NPs may play a crucial,or even a decisive role in catalytic reactions,despite catalytic reactions proceeding on the shell of nanoparticles.
In this article, alumina supported Ag@Pd NPs with Pd-shell thickness from 2 to 6 atomic layers upon a 2.6 nm Ag core were synthesized by a facile successive reduction method.Characterization of the catalysts was achieved by X-ray diffraction(XRD),transmission electron microscopy (TEM), Fourier-transform infrared(FTIR) adsorption spectroscopy of CO, and X-ray photoelectron spectroscopy (XPS), etc.Acetylene selective hydrogenation in ethylene-rich stream was performed to detect the catalytic properties of different Pd surfaces,including monometallic Pd,PdAg alloy and Ag@Pd core-shell with varing Pd/Ag atomic ratios.The kinetic measurements and DFT calculations were also conducted to help us to elucidate the dynamic evolution of Ag/Pd interface and shell-thickness-dependent catalytic performance.
2.Experimental
2.1.Materials
Silver nitrate (AgNO3, ≥99.8%), palladium acetylacetone(Pd(acac)2, ≥99.9%), N, N-dimethylformamide (DMF, ≥99.5%), and oleic acid (OA, 80%-90%) were purchased from Sigma-Aldrich,chemical grade, and used directly without further purification.Al2O3was purchased from Sasol Co.and calcined at 700 °C before use.
2.2.Catalysts preparation
In this work, Al2O3supported Ag@Pd NPs (1% Ag loading) with Pd/Ag molar ratios of 0.5:1, 1:1, and 2:1 were prepared by continuous reduction method using DMF as reducing agent.The Ag@Pd/Al2O3sample names were based on their target Pd/Ag molar ratios.Taking Ag@Pd-1 as an example, first, a certain amount of AgNO3(16.0 mg) and 10 ml OA were dissolved in 30 ml DMF and sonicated for 5 min to form a homogeneous solution.Then the solution temperature was increased to 140°C and kept for 1 h in an oil bath under stirring.After that,20 ml DMF solution containing Pd(acac)2(28.8 mg)was dropped into the mixture within 1 h and maintained for another 0.5 h.The resulting mixture was cooled to room temperature and 1 g Al2O3powder was poured into the flask with vigorous stirring for 4 h.Finally, the suspension was centrifuged and washed thoroughly with ethanol, followed successively by freeze-dry (-80 °C) overnight.The Ag@Pd-0.5, and Ag@Pd-2 samples were prepared in the same process except for the different amounts of Pd contents.Monometallic Ag/Al2O3and Pd/Al2O3catalyst with 1%(mass)metal loading was synthesized using the same method.Alumina supported PdAg alloy catalysts with varied Pd/Ag molar ratios were prepared by co-reduction of Ag and Pd precursors in the mixture of DMF and OA (10 ml OA in 50 ml DMF).The reaction temperature was increased to 160 °C and kept for 1 h under stirring condition.Following load,wash,and dry process are same as the preparation of Ag@Pd-1.The obtained PdAg alloy catalysts were denoted as PdAg-x (x = 0.5, 1, and 2).
2.3.Characterizations
UV-vis spectra were collected on a UV-2501PC spectrophotometer (SHIMADZU).XRD pattern of NPs and supported samples was recorded on a Rigaku Miniflex powder X-ray diffractometer coupled with a Cu Kα radiation source (λ = 0.15418 nm).To get XRD testing samples,the colloid of NPs was dispersed in a mixture of hexane and acetone, then centrifuged at 12,000 r·min-1for 10 min.The obtained slurry was dried on a glass slide in a 60 °C oven.The morphology and size of as-prepared Ag@Pd NPs were examined by transmission electron microscopy (TEM) and highangle annular dark-field scanning transmission electron microscopy(HAADF-STEM)equipped with energy dispersive X-ray spectroscopy (EDX), performing on a FEI Titan Themis microscopy,operating at 200 kV.More than 300 particles in different regions are randomly selected to measure the size distribution of Ag@Pd NPs.H2pulse chemisorption was taken to calculate the dispersion of Pd atoms (Micromeritics ASAP 2020).Around 0.1 g sample was pretreated at 120°C in diluted hydrogen and then cooled to 100°C using helium.The dispersion is determined on the assumption of a unit adsorption stoichiometry(H/Pd=1).The X-ray photoelectron spectroscopy (XPS) was conducted on an Axis Supra spectrometer and charge neutralizer with Al Kα radiation (1486.6 eV).Spectrum image data sets from an area of 800 μm × 800 μm, were acquired from C 1s, Pd 3d and Ag 3d photoelectron regions, at 50 eV pass energy, with base pressure in the instrument <10-7Pa.The binding energy of tested elements was calibrated using the energy of the C 1s peak (284.6 eV) as a reference, and spectra treatment has been performed using CASA software.
2.4.Catalytic testing
Acetylene hydrogenation was conducted in a fixed-bed quartz tube (9.525 mm OD) reactor.The reaction was carried out in the temperature range of 30-120 °C with 100 mg of catalyst diluting with 1 ml quartz sand.The reaction was operated at 0.4 MPa pressure with a mixture of 0.33%(vol)C2H2/33%(vol)C2H4/0.65%(vol)H2/balance N2,which simulated tail-end hydrogenation conditions.The total flow rate of the input gases was set to 166.5 ml·min-1with a space velocity of 10050 h-1.Before collecting data, the catalysts were pre-reduced at 200°C for 0.5 h and then cooled to room temperature.The outlet gas products were analyzed by an online GC with a flame ionization detector with a PLOT capillary column(0.53 m×50 mm).Negligible amounts of C4species and oligomers were detected during the short contact time among the hydrogenation process,and the total carbon deposition within long time testing was measured by thermos-gravimetric analysis.Acetylene conversion, ethylene selectivity and TOFs were calculated as follows:
where A is moles of acetylene conversion per second;n is Pd moles in the catalyst; D is metal dispersion.
2.5.Computational methods
All DFT(density functional theory)calculations were performed using the VASP code within periodic plane wave framework.The electron exchange and correlation terms were described with the PBE functional [33].The projector-augmented wave (PAW) pseudopotentials[34]were used to model the electron-ion interactions with an energy cutoff of 450 eV.The metal surfaces were modeled with slabs consisting of 8-layer thick 4 × 4 × 1 supercells, separated by a vacuum space of 1.6 nm in the z direction to eliminate interactions between successive slabs.The first Brillouin zone was sampled with a 3 × 3 × 1 Monkhorst-Pack k-point mesh[35].The DFT-D3 method was adopted to describe dispersion interactions [36].Only the bottom two layers was kept fixed during geometry optimizations, convergence criterion of which was a maximum force of 0.02 eV·10-10m-1.All transition states were located by the climbing image nudged elastic band method (CINEB) [37,38] with a convergence criterion of 0.05 eV·10-10m-1.In this work, we modeled four kinds of metal surfaces: Pd(1 1 1)surface with optimized lattice constant of 0.394 nm; PdAg(1 1 1)alloy surface with an unity Pd/Ag ratio and an optimized lattice constant of 0.404 nm; expansively strained Pd(1 1 1) surface with Ag lattice spacing(0.415 nm)and PdAg lattice spacing,denoted as Pd4.15(1 1 1) and Pd4.04(1 1 1), respectively; Pd overlayers on Ag that mimics the Ag@Pd core-shell catalysts, denoted as Ag@PdxML(x = 2, 4, 6; indicating the number of Pd overlayers).Structures of these metal surfaces are presented in Fig.S1 (in Supplementary Material) Binding energies (BE) of adsorbates were referenced to energies of the corresponding stable gas-phase molecules, e.g.BE of H atom was referenced to half of the energy of gas-phase H2molecule.BE change (ΔBE) on different surfaces with respect to Pd(1 1 1) were calculated and split into two parts: BE change caused by lattice constant change (ΔBE_strain) and BE change caused by Ag alloying(ΔBE_ligand),indicating the so-called strain and ligand effect on binding strength of adsorbates.
3.Results and Discussion
3.1.Synthesis and characterization of Pd-Ag bimetallic catalysts
When using N,N-dimethylformamide (DMF) as reducing agent,the morphology,size,and the reduction kinetics of ions can be controlled by adjustment of synthesis conditions.Our previous work[13] clarified that the deposition rate between Ag+and Pd2+in DMF solution can be easily tuned by changing the reaction temperature and metal ion concentration, and well-defined Ag@PdAg core-shell structure can be obtained.In this work, alumina supported Ag@Pd-x NPs was synthesized using DMF as both the solvent and reductant with OA as a stabilizer, the schematic illustration of the synthesis process is described in Fig.1(a).In this route,the distribution and relative molar ratios between Ag and Pd could be controlled by adjusting the reduction order and precursor contents in the raw solution.According to the Pd/Ag molar ratios,the obtained NPs was named Ag@Pd-0.5, Ag@Pd-1 and Ag@Pd-2.Fig.1(b) shows the UV-vis absorption spectra of Ag and series Ag@Pd colloids, and a strong peak at 426 nm is observed for Ag NPs colloid [39].Comparing to the spectra of Ag colloid, the total disappearance of the plasmon absorption peak over Ag@Pd samples suggests a complete coating of Pd shell.
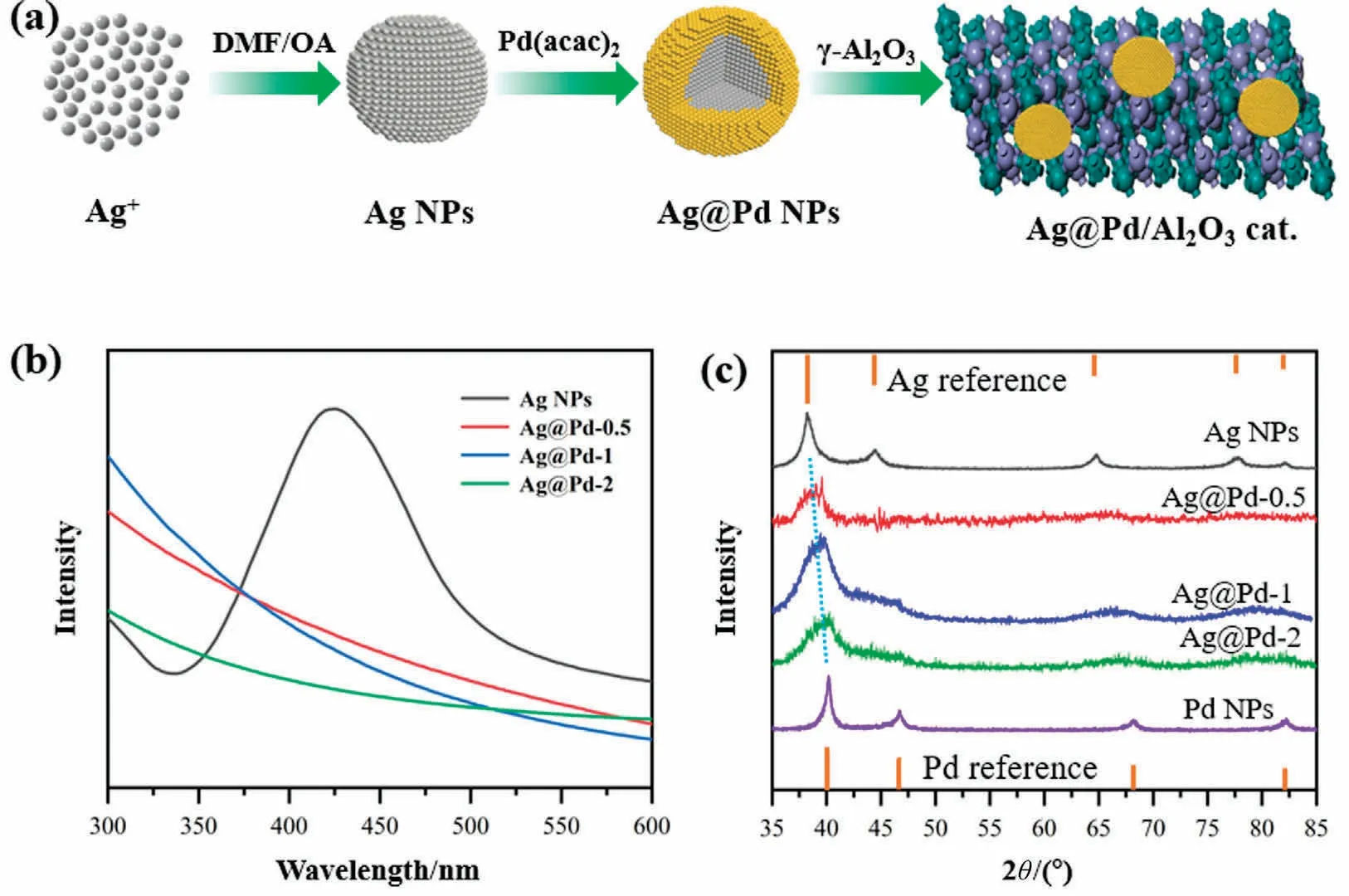
Fig.1.(a)Schematic illustration for preparation of the alumina supported Ag@Pd-x catalyst(x=0.5,1,and 2).(b)The UV-vis spectra of Ag and Ag@Pd-x colloidal solutions.(c) XRD patterns of the Ag, Pd and Ag@Pd-x NPs.
The coating of Pd on the Ag seeds also leads to other changes such as lattice parameter, as revealed by XRD.As shown in Fig.1(c), the XRD peaks recorded from the Ag NPs are located at 38.3°,44.2°, 64.5°, and 77.6°, which can be assigned to the (1 1 1),(2 0 0),(2 2 0),and(3 1 1)diffractions of Ag in face-centered cubic(fcc) phase, respectively.The peak positions of mono Pd NPs are consistent with the corresponding standard bulk Pd (fcc phase).The diffraction peak of (1 1 1) lattice plane over series Ag@Pd NPs are located between these for Pd and Ag,and these peaks shift to higher angle with increasing Pd content.Considering the lattice mismatch between Ag and Pd(0.408 nm vs.0.389 nm),two factors may contribute to this peak shift: (i) the formation of Pd-Ag alloy due to the inter-diffusion, resulting from galvanic replacement between Pd2+and Ag atoms, and (ii) the coating Pd shell on Ag seeds results in concomitant lattice contraction of Ag NPs crystal,and a further lattice contraction is detected with increasing Pd atomic layers.Previous studies demonstrated that oleylamine is able to protect the surface of Ag NPs and react with Pd(acac)2to form Pd(acac)x(OA)ycomplexes, preventing the redox reaction from each other [40-42].Thus, a relatively uniform Pd coating on the Ag surface can be achieved, leading to lattice contraction of Ag core.γ-Al2O3is taken to support as-prepared NPs, the XRD and BET analysis of Al2O3are showed in Fig.S2.
The size and morphology of NPs in colloid and supported catalysts were determined by TEM.As shown in Fig.S3, the size of Ag NPs is uniform and statistical analysis of Ag NPs displays that the average size of Ag NPs is 2.6 nm with high mono-dispersity(<±0.5 nm).Moreover, the Ag NPs distribute homogeneously on the alumina substrate after loading.The TEM images and corresponding size distribution of Ag@Pd-x NPs are displayed in Fig.2.It should be noted that seed-mediated growth to obtain Ag@Pd core-shell NPs without losing the regularity.Specifically, Ag@Pd-0.5,Ag@Pd-1,and Ag@Pd-2 all have a narrow size distribution and the corresponding mean diameter is 3.7, 4.5, and 5.3 nm, respectively.Coating Pd layers on Ag core increases the radius of NPs by 0.55, 0.95, and 1.35 nm, respectively.Approximating NPs as a sphere and 0.26 nm for atomic size of Pd atom, we can calculate that Ag@Pd-0.5, Ag@Pd-1, and Ag@Pd-2 possess around 2, 4, and 6 Pd atomic layers on Ag core.
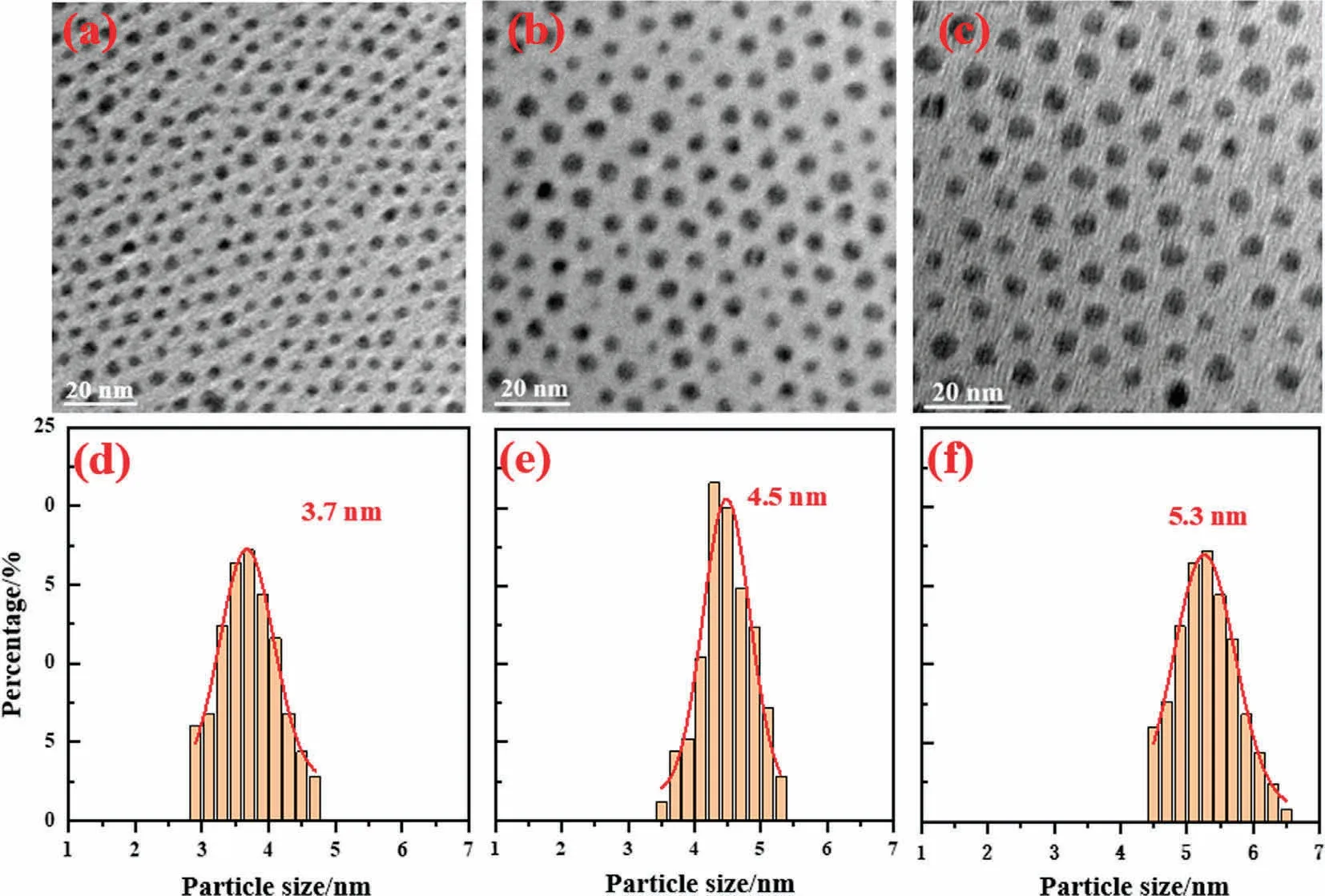
Fig.2.TEM images and corresponding size distribution of Ag@Pd-0.5 (a), (d), Ag@Pd-1 (b), (e), Ag@Pd-2 (c), (f).
High-angle annular dark-field scanning transmission electron microscopy coupled with energy-dispersive X-ray spectrosopy were taken to demonstrate the evolution of the Pd shell thickness for as-prepared Ag@Pd-x samples, as shown in Fig.3.Elemental mapping of Ag@Pd-0.5 clearly reveals that it comprised Ag NPs in the core and a nanolayer of Pd with a thickness of ca.0.5 nm in the shell,which is consistent with the calculated shell thickness based on the average size.Fig.3(b) shows the image of alumina supported Ag@Pd-2 nanoparticles, the lattice fringes with a spacing of 0.235 nm where observed in the core region, which is slightly smaller than that of the (1 1 1) diffraction plane of Ag(ca.0.238 nm)[43],in good agreement with the XRD analysis.Correspondingly, the shell region of Ag@Pd-2 NPs have the lattice fringe with a spacing of 0.226 nm, which is assigned to the(1 1 1) plane of Pd [44], indicating an expanded arrangements of surface Pd atoms.To further probe the elemental distribution of Ag and Pd, a corresponding EDX line profile was taken across the diameter of ~5.5 nm Ag@Pd-2 nanoparticle (Fig.3(c)), which clearly reveals the characteristic of core-shell structure with a thickness of ca.1.3 nm of Pd shell.This data is consistent with the results calculated from different average size between Ag and Ag@Pd-2 NPs.

Fig.3.(a)HAADF-STEM image and corresponding EDX elemental maps of Ag@Pd-0.5 sample.The length of the scale bars is 2 nm.(b)HR-TEM image and(c)EDX elemental line-scan of Ag@Pd-2 sample.
Electronic modification of Pd, arising from the addition of Ag atoms, was detected by XPS analysis.In order to distinguish the ligand effect originating from core-shell structure with different shell thickness, the electronic property of PdAg alloy with corresponding Pd/Ag ratio was also analyzed.As shown in Fig.S4,comparing with mono-Pd sample (335.0 eV), alloying Pd with Ag caused a clear shift to lower binding energies in Pd 3d5/2over PdAg-2 (334.9 eV), PdAg-1 (334.7 eV), and PdAg-0.5 (334.6 eV).The perturbation of Pd binding energy through the formation of Pd-Ag bond arise from the difference electronegativity of two metals, getting stronger with decreasing Pd/Ag ratios, which is in line with literature findings [45].Fig.4 shows the Pd 3d and Ag 3d XPS spectra of the fresh Ag@Pd-x catalysts, and only metallic Pd0(335.0 eV) and Ag0(367.7 eV) species are detected in these samples after pre-reduction.The Pd 3d5/2binding energies of the Ag@Pd-2 catalyst is located at 334.9 eV, nearly identical to that of mono-Pd sample.Further decreasing the Pd layers caused considerable shifts to lower binding energies up to 0.7 eV for the Ag@Pd-0.5 catalyst, suggesting that the influence of electronic property on surface Pd atoms increase sharply with declining Pd overlayers.The BE of Ag 3d5/2over Ag@Pd-x catalysts shift to higher BE (up to 0.3 eV) is indicative of electronic modification between Ag core and Pd shell.This electron interaction between Pd and Ag is more sensitive to Pd/Ag ratio in core-shell structure contrast to alloy structure.
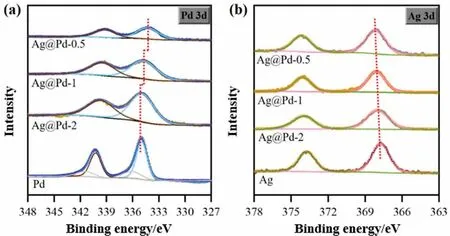
Fig.4.Pd 3d and Ag 3d XP spectra of alumina supported Ag@Pd-x samples and mono-Pd and Ag as a comparison.
3.2.Catalytic performance evaluation
Selective hydrogenation of acetylene to ethylene in ethylenerich atmosphere was performed to assess the catalytic properties of core-shell structure.The Ag/Al2O3sample shows negligible acetylene conversions at the temperature range of 20-100 °C.While keeping the catalyst dosage at 0.1 g, we measured the C2H2conversion and C2H4selectivity of Ag@Pd-x catalysts with increasing temperature, as displayed in Fig.5(a) and (b).These Pd-based catalysts are quite active, leading to full conversion of C2H2below 80 °C.It can be seen that the complete consumption of acetylene over Ag@Pd-x samples are achieved at lower temperature,with comparison to mono-Pd catalyst(100%C2H2conversion at 75 °C).At low conversions, mono-Pd catalyst shows acceptable olefin selectivity.However, the reaction product rapidly shifts towards C2H6near full conversion,and only 28%ethylene selectivity can be maintained.As for Ag@Pd-x samples,the curves of C2H4selectivity versus C2H2conversion exhibit a characteristic of slower decline.Noteworthily, Ag@Pd-x with different Pd shell thickness possess discrepant acetylene conversion and ethylene selectivity,even at same testing temperature.The highest ethylene selectivity is observed on Ag@Pd-1 catalyst, which exhibits 76% olefin selectivity at 65 °C, with >99% C2H2conversion.Thus, the activity and products distribution are influenced by both the subsurface composition and shell thickness.
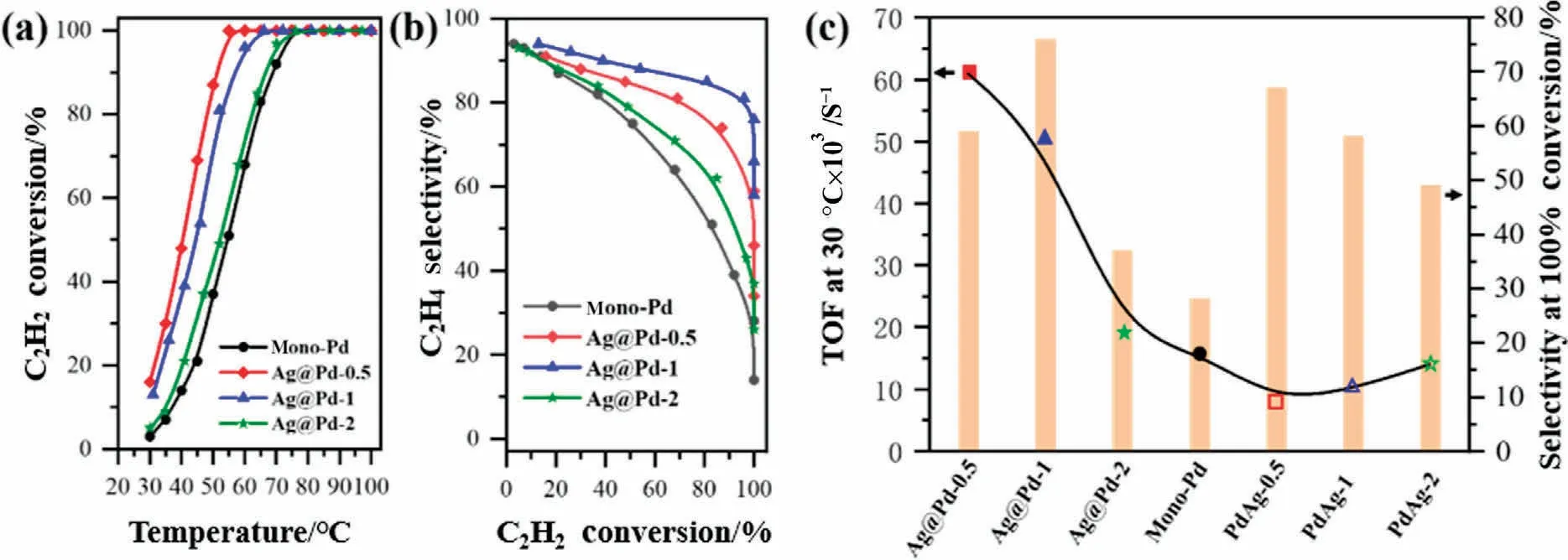
Fig.5.(a) Acetylene conversion as a function of test temperature and(b) ethylene selectivity as a function of the acetylene conversion of mono-Pd and Ag@Pd-x core-shell catalysts.(c) Catalytic activity and selectivity of the differently structured Pd-Ag catalysts in the selective hydrogenation of acetylene.
Normally, alloying Pd with a less active but more selective metal like Ag and Cu,such deep-hydrogenation can be partly suppressed, but not without compromising activity compared to pure Pd.To demonstrate the effect of the metal distribution on the catalytic properties,acetylene hydrogenation over alumina supported PdAg-x (x = 0.5, 1, and 2) alloy catalysts were also performed, and the plots of C2H2conversion and C2H4selectivity with increasing temperature were expounded in Fig.S5.Compared to mono-Pd catalysts, the conversion profiles of Pd-Ag alloy catalysts shift to higher temperatures, while a higher olefin selectivity can be obtained.In Fig.5(c),we show the catalytic activity at the reaction temperature of 30 °C and the C2H2selectivity at >99% acetylene conversion of all catalysts, to clearly display the different catalytic performance of core-shell and alloy structure.We compared the performance of different supported Pd-based catalysts in selective hydrogenation of acetylene, as shown in Fig.S6.Compared with other catalysts with complete acetylene conversion performance,Ag@Pd-1 catalyst in this paper can achieve complete acetylene conversion under mild conditions and maintain high selectivity.In addition,due to the unique core-shell structure of this catalyst,the active component Pd is located in the shell layer, so the same performance can be achieved with less precious metal consumption than alloy structure or mono-metal Pd catalyst.The catalytic activity is given as a turnover frequency (TOF), which is defined as the number of reacted C2H2molecules per second of each surface Pd atoms.The details of Pd dispersion and calculated TOF at 30 °C of each sample are available in Table 1.The Pd/Al2O3gives a TOF of 0.016 s-1, which matches the range of 0.01-0.03 s-1over reported Pd/Al2O3catalysts.The TOFs of three Pd-Ag alloy catalysts with different Pd/Ag ratios are smaller than mono-Pd catalysts,and the TOFs of alloy catalysts increase with increasing the Pd/Ag ratios.This result can be explained by the change in the nature of active site on the diluted Pd surface of PdAg alloy NPs, leading to an increasing of apparent activation energy from 41.9 to 54 kJ·mol-1, as displayed in Fig.6(a) and Table 1.Evidently,core-shell catalysts achieves higher activity than Pd-Ag alloy and Pd counterpart, especially the TOFs of Ag@Pd-0.5 and Ag@Pd-1 are up to 4 and 3 time higher than pure Pd catalyst,suggesting that the catalytic properties is significantly affected by the core NPs although the catalytic reaction takes place on the shell surface.Moreover,the variation trend of TOFs with increasing Pd/Ag ratios is different between core-shell and alloy structures.Specifically,the values of TOF decrease from 0.062 to 0.019 s-1with increasing Pd shell thickness over core-shell catalysts, while a higher TOF is observed with enhancing Pd/Ag ratios for alloy catalysts.Additionally, Ag@Pd-1 catalyst gives satisfactory stability, in which a constant percentage of C2H2conversion and C2H4selectivity is observed within a 28 h catalytic evaluation test (Fig.6(b)).
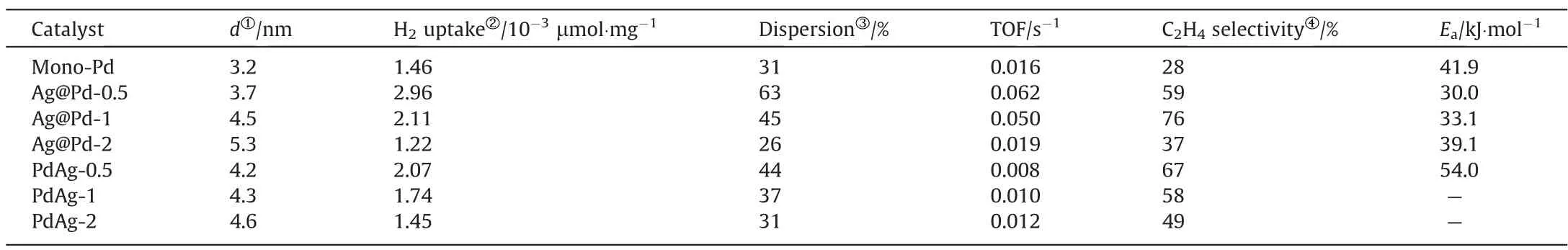
Table 1 The physicochemical properties of supported mono-Pd and bimetallic Pd-Ag catalysts
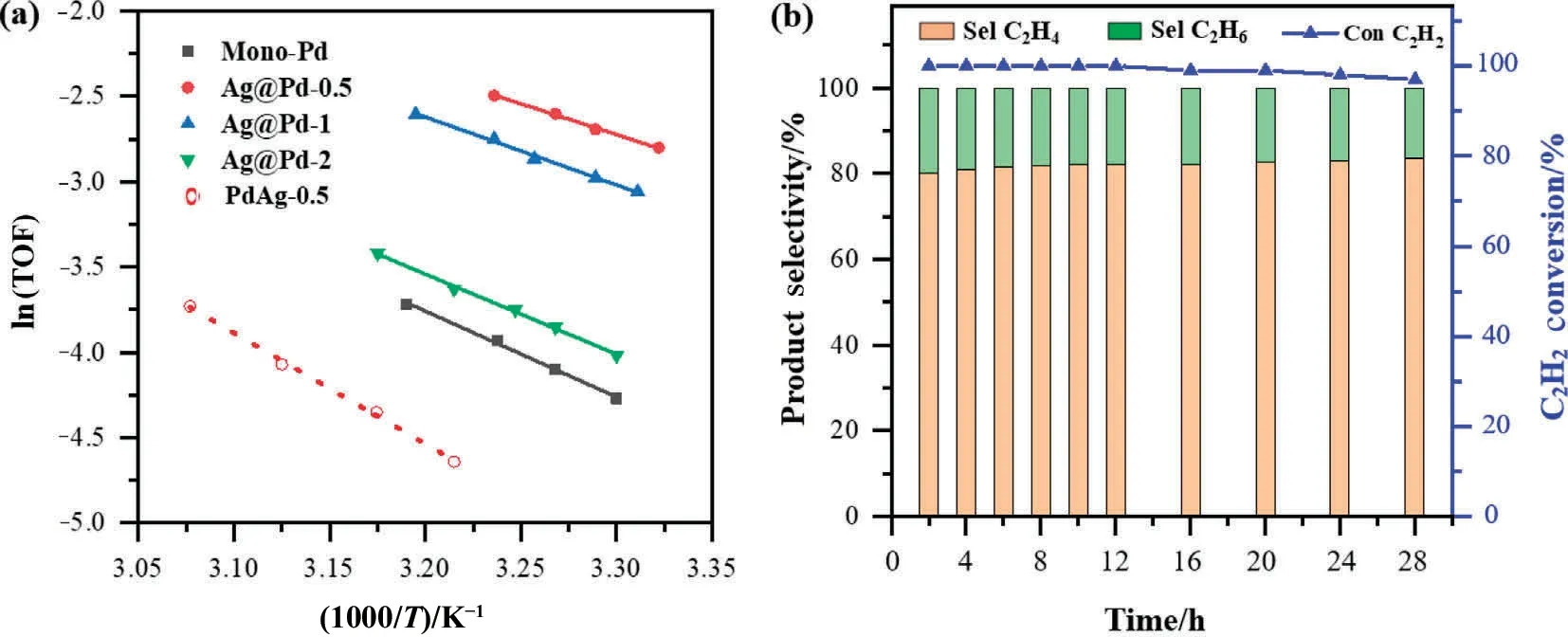
Fig.6.(a) Arrhenius plots of catalytic activity of mono-Pd, Ag@Pd core-shell, and PdAg-0.5 alloy catalysts.(b) Acetylene conversion and products selectivity versus testing time at 65 °C over Ag@Pd-1 catalyst, and the initial acetylene conversion is 99%.
3.3.DFT calculations on core-shell and alloy Pd surfaces
To understand the different catalytic performance of core-shell Ag@Pd and the uniformly distributed PdAg alloy catalysts,we performed density functional theory calculations.Binding modes of acetylene on Pd surfaces are apparently affected by lattice spacing:it prefers a η2η2coordination mode on unstrained Pd(1 1 1) and Pd4.04(1 1 1) undergoes 2.5% tensile strain, as lattice strain increases to + 5.3%, a η3η3coordination mode is more favorable on Pd4.15(1 1 1) and Ag@PdxMLsurfaces (Fig.7(a)).Due to the unique ensembles on PdAg(1 1 1) surface, acetylene adopts a different η1η1type coordination to two adjacent Pd atoms.Binding strength of acetylene increases as lattice expands: |BE| increases from 2.48 eV on Pd(1 1 1) to 2.52 eV and 2.62 eV on Pd4.04(1 1 1)and Pd4.15(1 1 1)surfaces,respectively(Figs.7(b)and S6).The three Ag@PdxMLall bind acetylene more strongly than Pd(1 1 1) by 0.1-0.5 eV.This binding strength change is mostly caused by ligand effect from Ag alloying in Ag@Pd2ML,which observes the strongest binding of acetylene among all surfaces.As Pd shell grows thicker beyond 4 monolayers (ML), ligand effect almost vanishes and strain effect starts to dominate the acetylene adsorption on Ag@PdxML(Fig.7(c)), resulting in similar C2H2BEs as on strained Pd4.15(1 1 1)surface with the same lattice spacing(Fig.7(b)).Compared to Pd(1 1 1),acetylene binds less strongly on the(1 1 1)surface of PdAg alloy catalyst by a significant 0.8 eV.In this case,ligand effect alone almost accounts for all the BE change, while strain effect is negligible.Deduced from the binding strength of acetylene, acetylene hydrogenation activity should follow:Ag@Pd2ML>Ag@Pd4ML~Ag@Pd6ML>Pd(1 1 1) >PdAg, in good agreement with the experimentally measure TOF trend and Ea.
We further investigated the deep-hydrogenation reaction from ethylene to ethane to understand the selectivity governing factors.Selectivity towards ethylene is determined by competition between ethylene desorption and its further hydrogenation to ethane.The binding strength of ethylene is also affected by strain and ligand effect on core-shell and PdAg alloy catalyst, which is in a similar trend with acetylene (Fig.S8).Ligand effect plays a dominant role in mixed PdAg alloy catalyst and Ag@Pd core-shell catalyst with shell thickness thinner than 2ML, then diminishes sharply and nearly vanishes beyond 4ML with increasing shell thickness.Specifically, ethylene prefers a di-σ binding mode(Fig.S9) on all the Pd and PdAg alloy surfaces.Compared to Pd(1 1 1), Ag alloying in PdAg weakens ethylene adsorption by 0.31 eV, Ag@Pd2MLstrengthens ethylene adsorption by 0.16 eV,while all other Pd surfaces show insignificant variance regarding the C2H4binding strength (Table S1).Based on the calculated potential energy diagram of ethylene hydrogenation (Fig.8(a)),the deep hydrogenation to ethane is rate-limited by the 2nd hydrogenation step (via a C2H5-H complex, inset in Fig.8(b)) on all three kinds of Pd surfaces.Therefore, activation energy of C2H4hydrogenation is calculated from adsorbed C2H4to the 2nd deep-hydrogenation transition state (TS), and we take the energy difference (δE) between the activation energy and C2H4desorption energy as the indicator of ethylene selectivity(δE=Eactivation-Edesorption,Fig.8(b)).Using the derived BEP scaling relations for C2H4hydrogenation (Fig.S10), we managed to correlate the selectivity descriptor δE with a lumped term of linear combinations of BE(H) and BE(C2H4) with great linearity (SA1 and Fig.S11).This lumped term[0.5BE(C2H4)-BE(H)],therefore,can be taken as the selectivity descriptor and neither BE(H) nor BE(C2H4)alone can be used as a good selectivity descriptor.
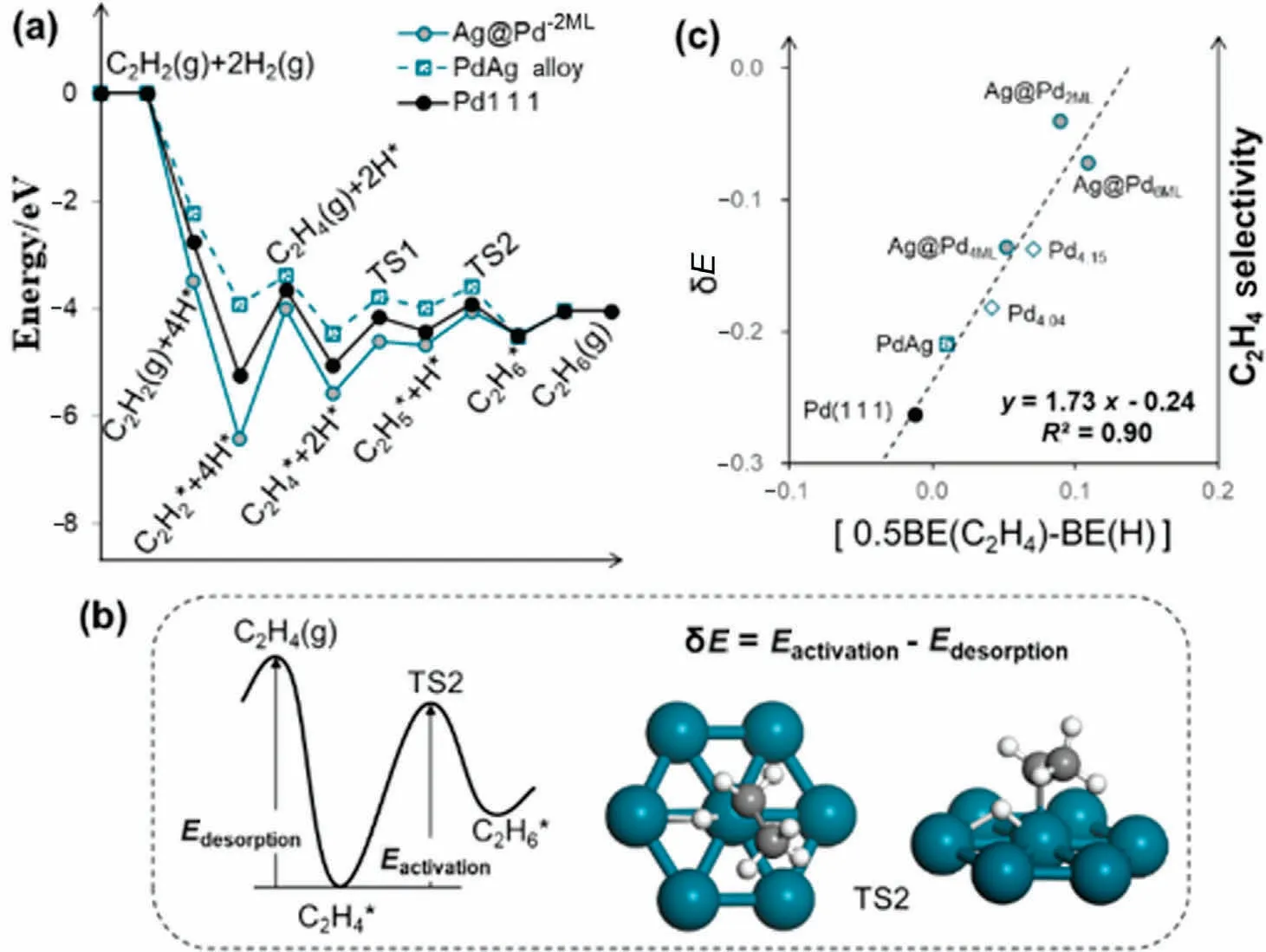
Fig.8.(a)Potential energy diagram of acetylene hydrogenation to ethane on Pd(1 1 1),PdAg(1 1 1)and Ag@Pd2ML.(b)Illustration of the calculation of selectivity indicator δE.(c) Correlation between δE and a lumped BE term [0.5BE(C2H4)-BE(H)] as selectivity descriptor.
Because of the similar BE changes with respect to Pd(1 1 1) for C2H4and H(Table S1)and the higher coefficient associated with BE(H) in δE, BE(H) plays a more important role than BE(C2H4) in determining ethylene selectivity during acetylene hydrogenation.All strained Pd surfaces explored in this work bind H more strongly than unstrained Pd(1 1 1), therefore we predict a higher ethylene selectivity on those strained Pd surfaces.Compared to unstrained Pd(1 1 1), PdAg alloy reduces the ethylene binding strength to a much higher extent than H.As a result,ethylene selectivity in this case is more determined by BE(C2H4),with a more positive value of which on PdAg than on Pd(1 1 1), we again predict a higher ethylene selectivity on PdAg.Our predicted selectivity trend(Fig.7(c)) Ag@PdxML>Pd4.15>Pd4.04>PdAg >Pd(1 1 1) from calculations is does not perfectly match the experimentally observed one.One possible reason could be our catalyst model do not accurately represent the real catalyst used in experiments.For example, the Pd shell is stretched to match the exact lattice constant of Ag in our core-shell Ag@Pd model.However, it is very likely the Pd shell is stretched, meanwhile the Ag core is compressed to reach a comprise in real situation.Other reasons include catalyst segregation might happen to some extent, defect sites could play a role or surface coverage of reactive intermediates may be high enough not to be neglected,etc.Despite the disparity between simulated selectivity trend and experimental trend, it does not diminish the validity and predictive power of the reactivity descriptor we derived.In acetylene hydrogenation reaction,not only the BE of C2H4but also the BE of H determines the ethylene selectivity, which can be accurately and easily deduced from the two BEs.The analysis we presented here in finding selectivity descriptors could be applied to other catalytic systems as well.
4.Conclusions
In this article, Ag@Pd core-shell NPs with controllable Pd shell thickness (0.55-1.35 nm) and PdAg alloy with same Pd/Ag ratios were synthesized to discuss the specific synergetic mechanisms arising from Ag addition, but with different nanoparticle surfaces.Combing the results from characterizations and kinetic measurements of acetylene hydrogenation, we found a clear Pd shellthickness-dependent performance in this reaction.In a brief,engineering Pd-shell surface could lower apparent activation energy,and Ag@Pd-0.5 catalyst exhibited 3-6 times more active than both pure Pd and PdAg alloy surfaces, following hydrogenation activity order:Ag@Pd2ML>Ag@Pd4ML~Ag@Pd6ML>Pd(1 1 1)>PdAg.Theoretical calculation results suggest that the ligand effect plays the major role on binding strength of acetylene for Ag@Pd structure,and this effect diminishes sharply with increasing Pd overlayers.Concomitantly, strain effect starts to dominate the acetylene adsorption.As for ethylene selectivity, the lumped term [0.5BE(C2H4)-BE(H)] was established to describe the selectivity changes of ethylene on different surfaces, which show a good linear relationship with ethylene selectivity indicator (δE = Eactivation-Edesorption).The exploration of dynamic evolution of synergetic mechanisms among bimetallic NPs with varied composition in this work can help us to deepen the understanding catalyst structureactivity relationship and provide a new perspective for the rational design of other catalytic systems.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by National Key Research&Development Program of China (2022YFA1506200), the National Natural Science Foundations of China (22078007, 21627813, 21706009,22002085), Guangdong Basic and Applied Basic Research Foundation (2020A1515110832), the Fundamental Research Funds for the Central Universities (buctrc201921, JD2223), Innovative Achievement Commercialization Service-Platform of Industrial Catalysis, MIIT.R.M.would like to thank Chemistry and Chemical Engineering Guangdong Laboratory for a startup funding support(2111001).
Supplementary Material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.cjche.2023.06.012.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Intrinsic kinetics of catalytic hydrogenation of 2-nitro-4-acetylamino anisole to 2-amino-4-acetylamino anisole over Raney nickel catalyst
- Experiments and model development of p-nitrochlorobenzene and naphthalene purification in a continuous tower melting crystallizer
- α-Synuclein: A fusion chaperone significantly boosting the enzymatic performance of PET hydrolase
- Influence of water vapor on the separation of volatile organic compound/nitrogen mixture by polydimethylsiloxane membrane
- Mass transfer mechanism and relationship of gas-liquid annular flow in a microfluidic cross-junction device
- Enhanced photocatalytic activity of methylene blue using heterojunction Ag@TiO2 nanocomposite: Mechanistic and optimization study