全基因组关联分析揭示白羽肉鸡孵化性状的遗传基础
2023-02-27张高猛丁纪强刘昱宏郑麦青赵桂苹李庆贺
张高猛,丁纪强,刘昱宏,郑麦青,文 杰,赵桂苹,李庆贺
(中国农业科学院北京畜牧兽医研究所,北京 100193)
鸡肉是我国第二大肉类生产和消费品,以孵化性状为代表的繁殖性能越来越重要[1],然而长期以来,在白羽肉鸡繁殖性能的选育中,育种工作者更加注重母鸡的繁殖效率选择,导致公鸡繁殖能力的选择较少。而种公鸡繁殖力的优劣直接影响后代的生产性能和肉鸡养殖的经济效益。因此,研究与公鸡繁殖性状相关的候选基因对提高肉鸡生产的经济效益具有重要意义[2],家禽育种中,传统方法难以提高个体繁殖率,而数量性状基因座(quantitative trait locus, QTL)潜在候选基因的研究对重要经济性状影响很大。有研究利用GWAS方法分析了396只京海黄鸡核心群母鸡11个繁殖性状相关的SNPs[3],张涛等[4]利用GWAS方法研究了京海黄鸡繁殖性状相关的分子标记和候选基因,结果发现有7个SNPs与繁殖性状相关。由此可见,标记辅助选择(marker-assisted selection, MAS)可以对繁殖性状进行改良,从而提高家禽养殖的经济效益。尽管动物的QTL研究长达20多年[5],但对公鸡受精率和受精蛋孵化率的QTL定位研究还比较少。伴随着基因分型技术的不断发展,可以更好的从基因组水平和转录组水平解析受精率和受精蛋孵化率的遗传基础[6]。本研究基于白羽肉鸡A、B两个资源群体,使用本团队自主研发的“京芯一号”55K SNP芯片对A群体(556只)和B群体(398只)共954个个体进行基因分型,使用一般线性混合模型(LMM)对受精率和受精蛋孵化率进行全基因组关联分析(GWAS),筛选与孵化性状相关的SNP位点。
随着测序技术的不断发展,繁殖性状也被证明与基因表达水平有关[7]。有研究通过深度测序探究了卵巢、垂体和下丘脑miRNA与产蛋性能的关联[8],发现前列腺素D2和WNT信号转导途径相关的H-PGDS和WNT2在低精子活力表型公鸡睾丸中的表达水平较低[9]。本研究对A群体G9世代8只公鸡睾丸组织进行转录组测序,鉴定孵化性状的转录水平差异,进一步解析孵化性状的遗传基础,从而为后期的分子标记辅助选择奠定基础。
1 材料与方法
1.1 试验动物
A、B资源群体采用笼养方式饲养,常规免疫,各世代营养水平保持不变,为减少其他因素对公鸡孵化性状的影响,每只公鸡采精后,对12只母鸡进行人工授精,收集10 d种蛋后,经甲醛+高锰酸钾3倍量(即每立方米空间使用甲醛42 mL,高锰酸钾21 g)熏蒸20 min。第18天照蛋时统计12只母鸡的受精率后取均值,受精率为入孵蛋数中受精蛋所占的比例。21 d出雏时统计12只母鸡的孵化率后取均值,孵化率为出雏数占受精蛋数的比例。A资源群体5个世代分别有80、76、120、120、160只公鸡统计受精率和孵化率,B资源群体3个世代分别有118、120、160只公鸡统计受精率和孵化率。对每只公鸡翅静脉采血,用肝素钠抗凝管保存于-20 ℃,用于基因组DNA的提取。A群体G9世代公鸡400日龄时,根据受精率高低挑选8只公鸡,试验组和对照组各4只,解剖采样上述睾丸组织,置于液氮冻存,用于RNA的提取。
1.2 遗传力估计
使用R语言中的shapiro.test函数对受精率和孵化率表型进行正态检验,符合正态分布(P>0.05)。分别利用系谱信息和基因组信息构建亲缘关系矩阵,使用Asremlv4.1软件包分别计算受精率的遗传力,Wald F法估计世代和批次的效应(P<0.01),因此计算时将世代和批次作为固定效应进行矫正[10],BLUP模型如下:
y=Xb+Zα+ε
其中,y是表型值向量,X和Z是固定效应和加性遗传效应的关联矩阵,b是固定效应向量,α是随机加性遗传效应向量,假设α~N(0,G/Aσ2α),ε是随机残差向量,α~N(0,Iσ2e),G是全基因组标记构建的亲缘关系矩阵(G矩阵),A是基于系谱的血缘关系矩阵(A矩阵)。
1.3 SNP分型质控
使用常规的酚-氯仿法提取血液中的基因组DNA,-80 ℃保存,将质量合格的样品送至北京康普森生物技术有限公司,使用“京芯一号”鸡55K SNP芯片进行基因分型,使用plink(V1.9)软件对芯片基因型数据进行质控[11],剔除基因型缺失率大于10%的个体、最小等位基因频率小于5%、样本检出率小于90%和分型错误的SNP。
1.4 全基因组关联分析
采用GEMMA(V0.98.1)软件(https://github.com/genetics-statistics/GEMMA/releases)中的单性状混合线性模型对受精率、孵化率性状进行GWAS分析,该模型包括SNP作为固定因子和个体的亲缘关系作为随机效应[12],混合线性模型中加入批次作为固定效应,固定效应首先采用R(V3.6.0)软件中model.matrix()函数转换成0和1的设计矩阵,然后以类似于协变量的形式加入GEMMA软件中。统计模型如下:
y=Wα+xβ+u+;u~MVNn(0,λτ-1K),~MVNn(0,τ-1In)
公式中,y代表表型值向量,W代表协变量(固定效应)的设计矩阵,包括第一列为1,α代表包括截距的相关系数向量,x代表标记基因型向量,β代表标记效应,u代表随机效应向量,代表残差向量,τ-1代表残差的方差,λ代表两个方差组分的比率,K代表由SNPs估计的中心亲缘关系矩阵,In代表单位矩阵。MVNn代表n维多元正态分布。Wald检验用于筛选与性状相关SNP的标准。使用plink(V1.90b)软件中的参数-indep-pairwise 25 5 0.2来推断独立检验的有效SNP数量,最终推断出有效独立检验SNP为6 950个。因此全基因组水平显著阈值和全基因组水平潜在显著阈值分别为7.19×10-6(0.05/6 950) 和1.44×10-4(1/6 950)。
1.5 RNA提取、质控、文库构建和测序
采用氯仿法提取RNA,使用NanoDrop 2000微量分光光度计检测RNA浓度,Agilent 2100 Bioanalyzer,Aglient RNA 6000 Nano Kit检测RNA的样品浓度和完整性,提取的RNA样品浓度≥1 000 ng·μL-1,RIN值≥8且28S∶18S≥1.2。总RNA样品检测合格后,根据不同来源mRNA的特性进行纯化。真核生物mRNA 3′末端具有polyA尾结构,选用带有Oligo(dT)的磁珠进行富集纯化;原核生物mRNA不具有该特性,选用试剂盒去除rRNA以获取更多有效信息。向纯化得到的mRNA中加入Fragmentation Buffer使其片断成为短片段。再以片断化后的RNA为模板,利用随机引物进行逆转录过程,实现cDNA第一链合成,并加入2nd Strand Marking Buffer、2nd Strand/End Repair Enzyme Mix合成cDNA第二链。后经末端修复、加碱基A,加测序接头,经磁珠筛选回收目的片段,并进行PCR扩增,完成整个文库构建。文库构建完成后,先使用Qubit3.0进行初步定量,稀释文库至1 ng·μL-1,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Bio-RAD CFX96荧光定量PCR仪、Bio-RAD KIT iQ SYBR GRN进行q-PCR,对文库的有效浓度进行准确定量(文库有效浓度>10 mol·L-1),以保证文库质量。质量合格的文库用Illumina平台进行测序。为了保证后续分析数据的质量,对原始序列进行过滤,去除接头污染的Reads,低质量的Reads(Reads中质量值Q≤19的碱基占总碱基的50%以上),去除含N比例大于5%的Reads后,得到高质量的Clean Reads,再进行后续分析。
1.6 差异基因筛选及表达量分析
以A群体低受精率公鸡为对照,以Fold change(FC)≥1.5、P-value<0.05且P-adj<0.05作为筛选标准筛选差异基因,两组公鸡睾丸组织中基因表达水平的差异以及差异的统计学显著性通过火山图显示,FPKM[13](Fragments Per Kilobase of transcript per Million fragments mapped)是利用RNA-seq技术定量估计基因表达值的一个非常有效的工具,即每百万fragments(测序核酸片段,PE中一条fragments对应两条reads)中来自某一基因每千碱基长度的fragments数目,其同时考虑了测序深度和基因长度对fragments计数的影响,是目前最为常用的基因表达水平估算方法。采用FPKM作为衡量基因表达水平的指标,使用DESeq2软件分析样品间的差异表达。
1.7 实时荧光定量PCR验证
cDNA反转录参照FastKing一步法除基因组cDNA第一链合成预混试剂试剂盒(天根,北京)操作说明,反转录产物于-20 ℃冻存备用,RT-qPCR以β-actin为内参基因,根据NCBI GenBank中鸡THYN1、HMGCLL1、COA6基因和β-actin基因序列设计引物(表1)。扩增体系:2×ChamQ Universal SYBR qPCR Master Mix(Vazyme) 10.0 μL,上、下游引物各0.4 μL(10 μmol·L-1),cDNA模板2.0 μL,7.2 μL水补足20 μL体系。扩增程序:95 ℃ 30 s;95 ℃ 10 s,60 ℃ 30 s,40个循环。采用7500型荧光定量PCR仪(ABI,美国)对THYN1、HMGCLL1、COA6基因和β-actin进行分析,每个样品设置3个技术重复。采用比较Ct值法,即2-ΔΔCt法计算基因相对表达量,使用SAS 9.1.3软件对每个基因的相对表达量进行双尾非配对T检验,P<0.05表示差异显著,P<0.01表示差异极显著。

表1 RT-qPCR所用基因的引物信息
2 结 果
2.1 遗传力估计
使用ASREML v4.1软件包构建基于系谱信息的传统动物模型(A矩阵),基于基因型信息构建的GBLUP(G矩阵),分别计算受精率的遗传力。Wald F统计世代和批次的Wald显著(P<0.01)。基于A群体5~9世代系谱和556个个体受精率表型值计算得遗传力为0.21,基于G矩阵计算受精率遗传力为0.14。
2.2 表型和基因型数据质控
A群体556个个体受精率表型最大值和最小值分别为0.96和0.28、平均值为0.84、变异系数为9%;孵化率表型最大值最小值分别为1.00和0.68、平均值为0.88、变异系数为7.1%;B群体398个个体受精率表型最大值和最小值分别为0.95和0.44、平均值为0.79、变异系数为9.9%;孵化率最大值和最小值分别为0.98和0.60、平均值为0.85、变异系数为7.9%。质控条件:剔除个体基因型缺失率大于10%的个体、最小等位基因频率小于5%、样本检出率小于90%的SNP。质控后,A群体保留32 459个SNPs,平均标记密度为31.7 kb/SNP(表2);B群体保留38 150个SNPs用于后续分析,平均标记密度为27.0 kb/SNP(表3)。

表2 质控后SNPs标记在A资源群体各染色体上的分布
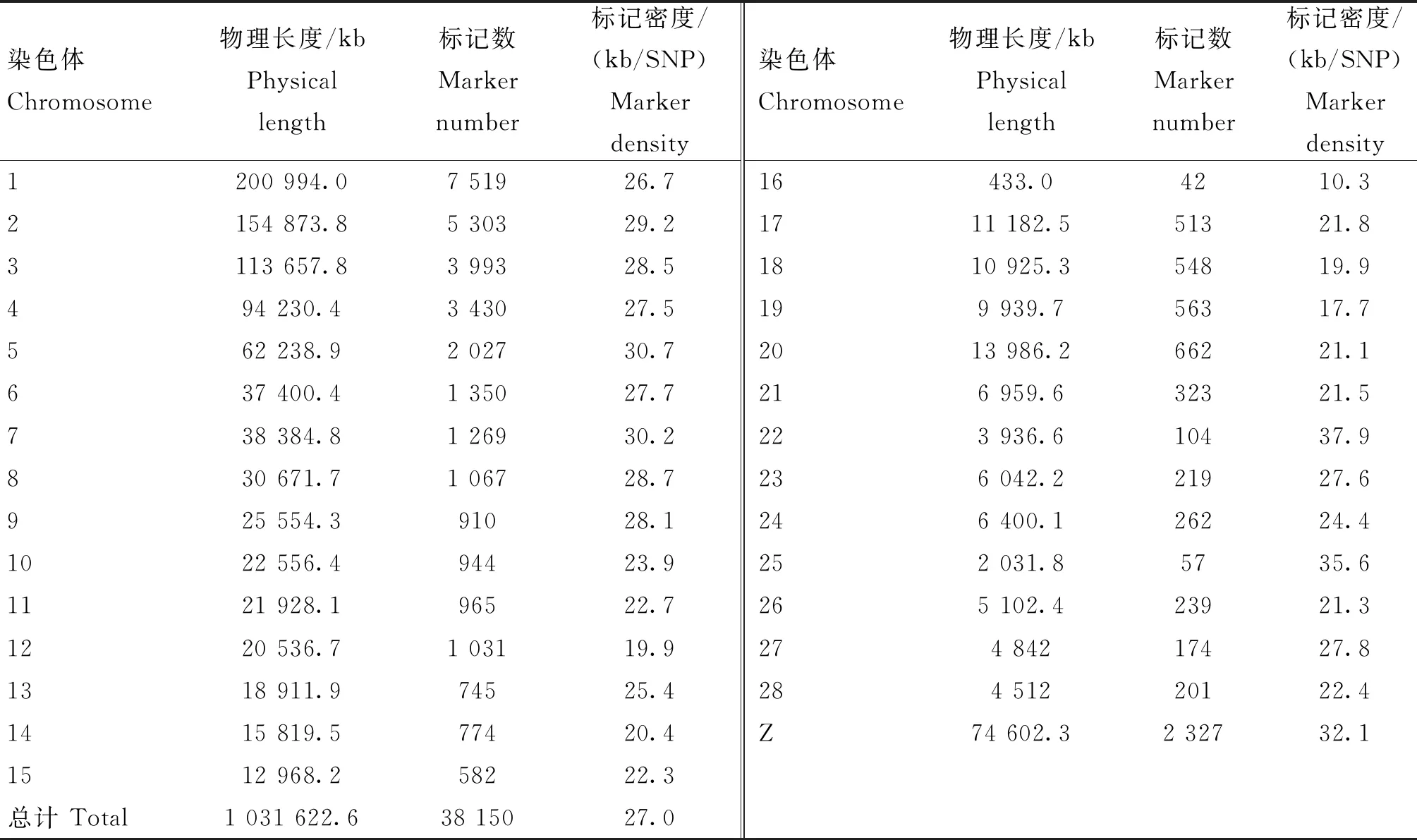
表3 质控后SNPs标记在B资源群体各染色体上的分布
2.3 主成分分析
PCA分析中,主成分1为横坐标,主成分2为纵坐标,以此做主成分散点图,由图1可知,两资源群公鸡各个世代没有聚拢在一起,说明群体之间出现分层。在以往的全基因组关联分析当中,由于祖代或者父母代的遗传差异导致的群体分层会导致分析结果出现假阳性[14]。利用全基因组标记的遗传信息对样本遗传相关进行估计,比系谱信息更能真实的反映了个体间的相关度,可以通过估计个体间的遗传相关来矫正群体结构分层对关联分析结果的影响,因此在关联分析中可以使用主成分1作为协变量,以校正群体结构对关联分析的影响[15]。

a.A群体结构主成分分析图;b.B群体结构主成分分析图
2.4 全基因组关联分析
受精率和孵化率的全基因组关联分析结果(图2)显示,有9个位点与受精率显著相关(P<1.01×10-4),其中A群体556只个体的受精率筛选到4个显著位点分别位于12号染色体的COPG1基因,11号染色体的ADAMTS18、CDH8基因,4号染色体的FABP2基因;B群体398只个体受精率筛选到4个显著位点,分别位于20号染色体的SLCO4A1基因,9号染色体的ATP13A3基因,1号染色体的NELL2、VEZT基因;A、B两群体基因型数据合并后,954只个体受精率筛选到1个显著位点,位于5号染色体的IGHMBP2基因。B群体398只个体孵化率性状筛选到3个显著位点(P<4.57×10-8),位于18号染色体的TMEM、SCO1、MYH1A基因。显著SNPs信息见表4。
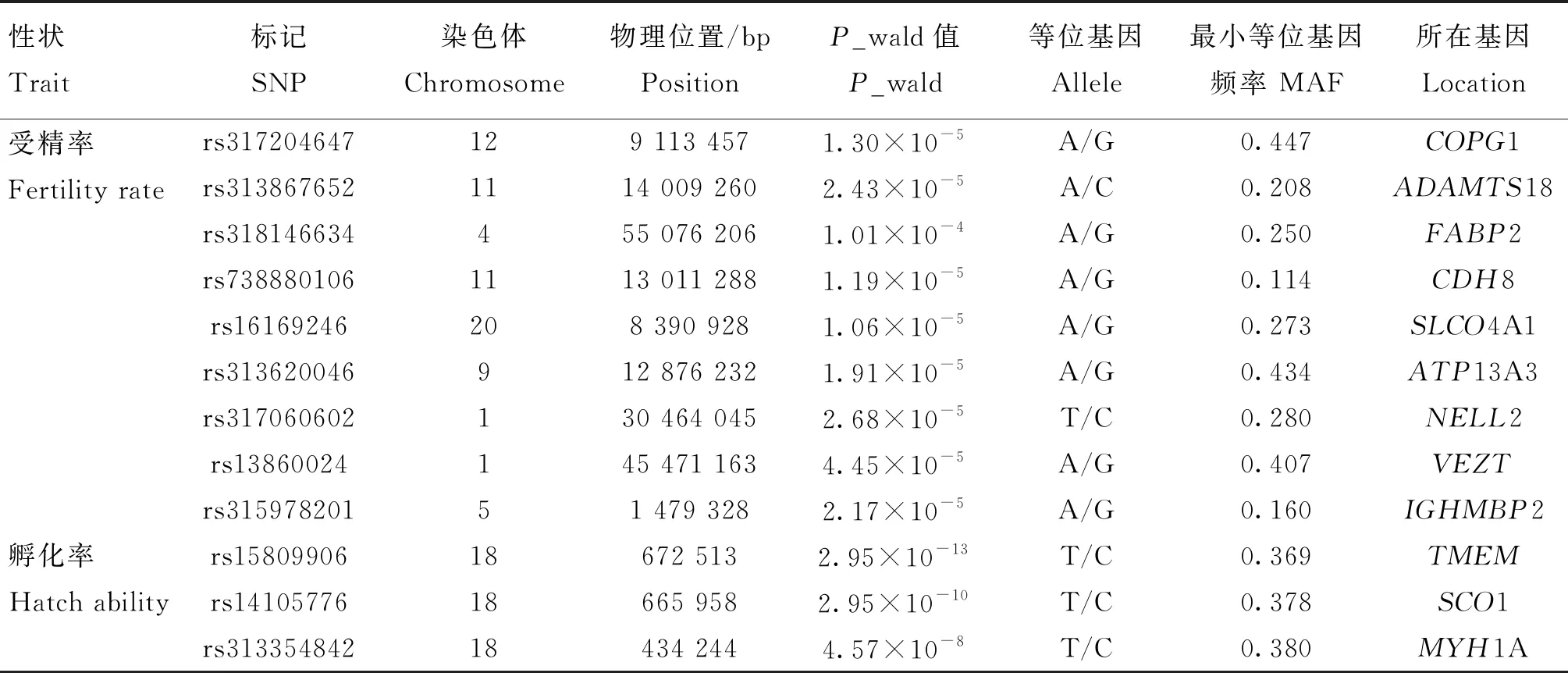
表4 受精率和孵化率达到5%基因组水平显著的SNPs位点信息
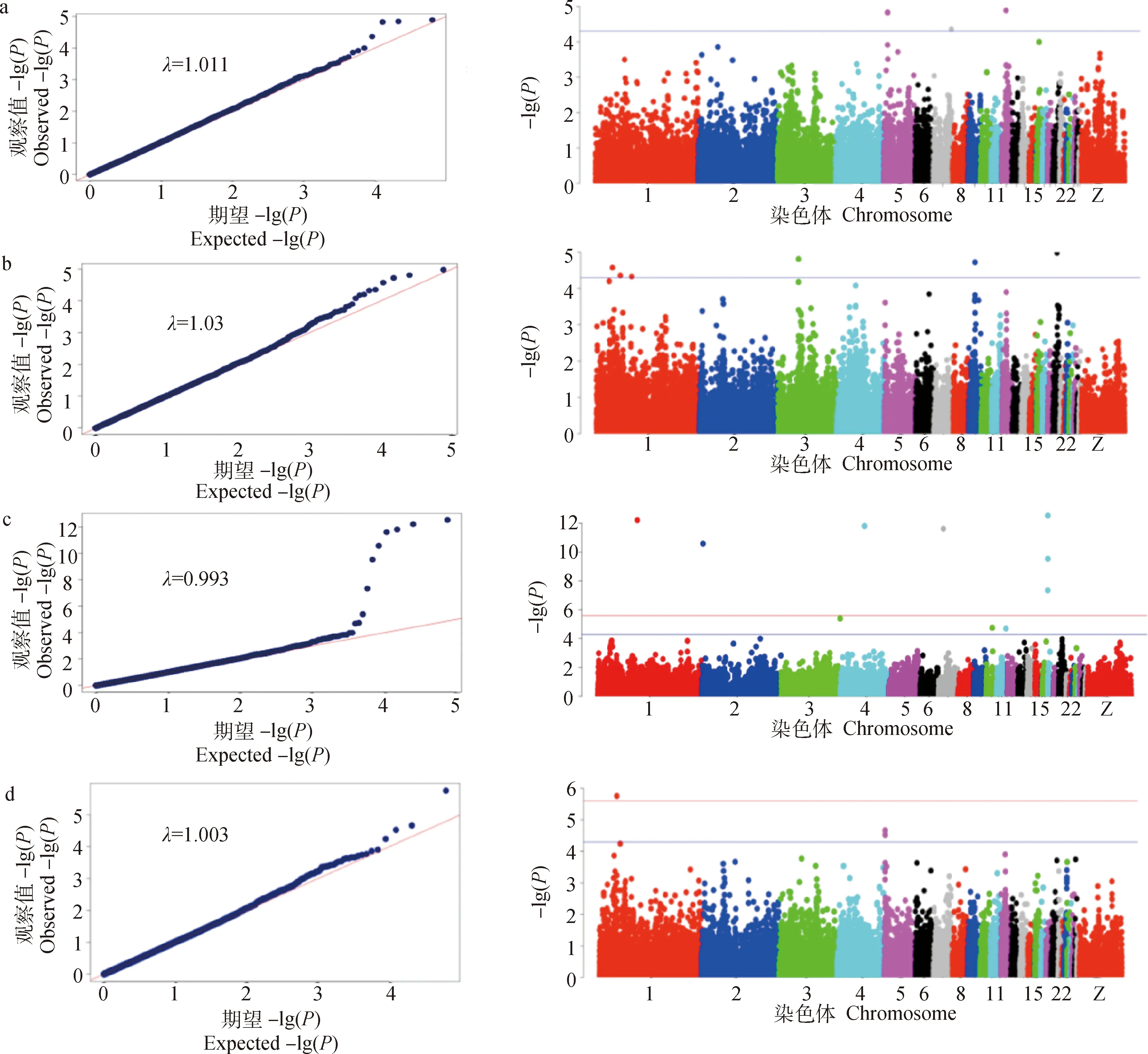
a.A群体受精率GWAS分析QQ图与曼哈顿图;b.B群体受精率GWAS分析QQ图与曼哈顿图;c.B群体孵化率GWAS分析QQ图与曼哈顿图;d.A、B群体芯片数据合并后受精率GWAS分析QQ图与曼哈顿图
2.5 转录组测序样本表型和测序分析结果统计
受精率高组4个个体受精率表型最大值和最小值分别为0.97和0.93、平均值为0.95;睾丸重最大值为47.34 g、最小值为24.08 g、平均值为36.3 g;受精率低组4个个体受精率表型最大值和最小值分别为0.85和0.79、平均值为0.83;睾丸重最大值为35.9 g、最小值为16.13 g、平均值为26.4 g。转录组测序分析结果见表5。
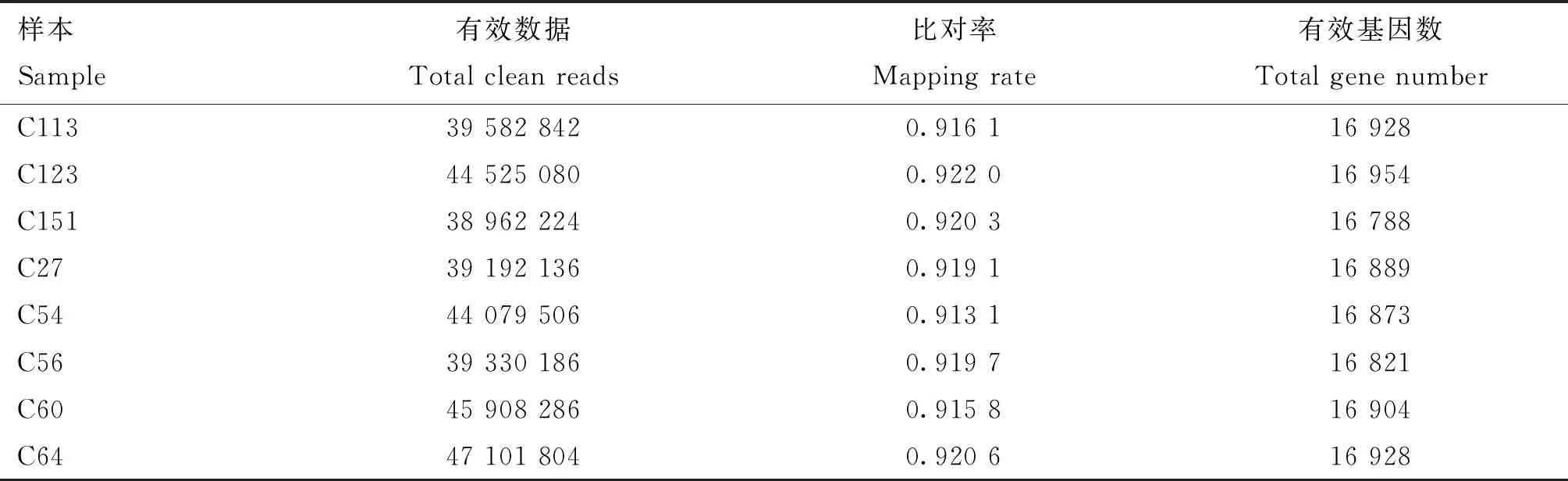
表5 8个样本转录组测序分析结果统计
2.6 差异表达基因筛选和差异表达分析
以白羽肉鸡受精率低组为对照组,以Fold change≥1.5、P-value<0.05且P-adj<0.05作为筛选标准,共筛选出差异表达基因17个,其中上调基因13个,下调基因4个。受精率高、低公鸡睾丸组织中基因表达水平的差异通过FPKM来定量估计,对受精率高、低组的白羽肉鸡差异基因进行表达量分析,发现低受精率组THYN1、TARBP1、ENSGALG00000036249、ENSGALG00000039805基因表达量较高,高受精率组HMGCLL1、COA6、ENSGALG00000033622基因表达量较高(图3)。
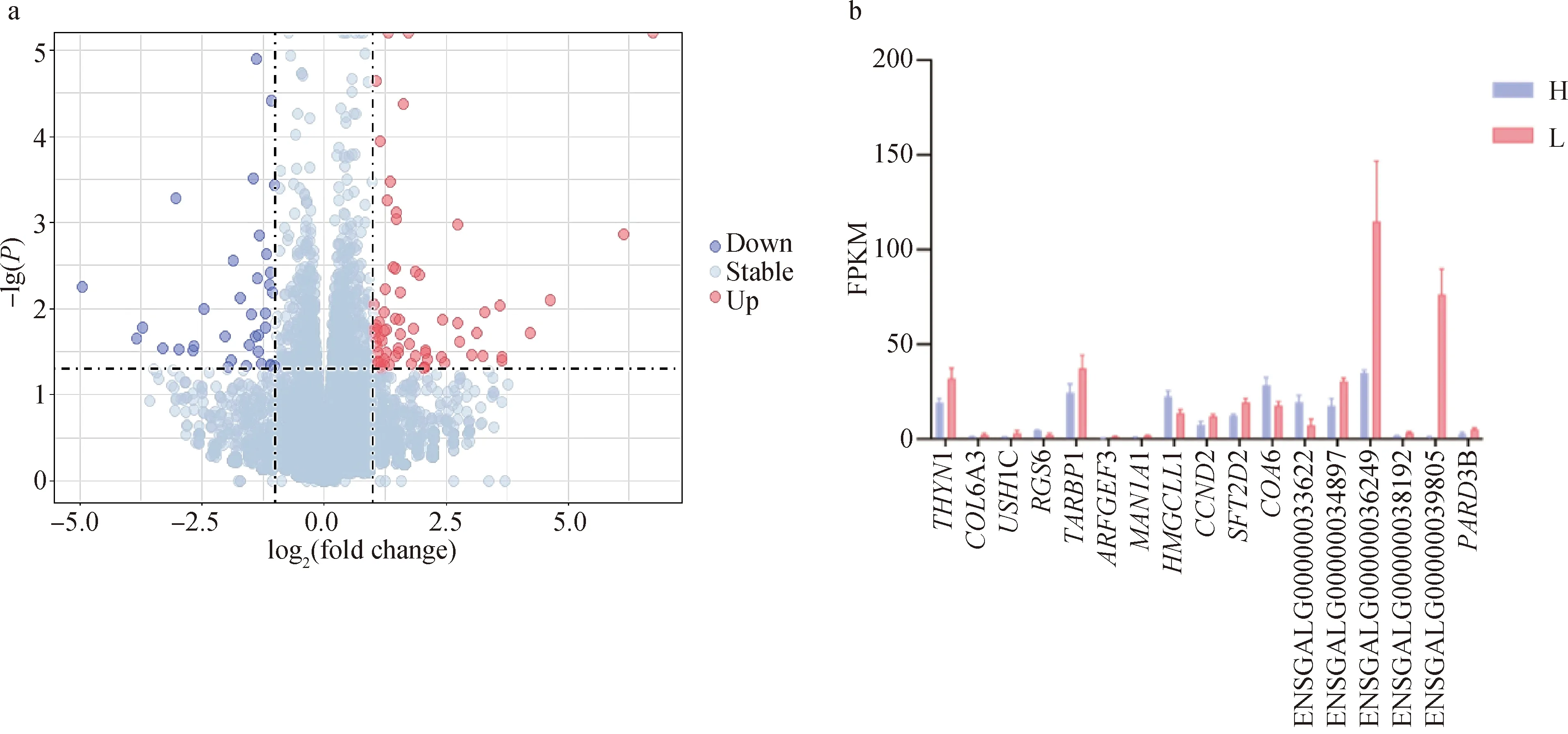
a.差异表达基因火山图;b.差异表达基因估计表达值图。L.受精率低组;H.受精率高组,下同
2.7 高、低受精率公鸡睾丸中HMGCLL1、COA6 mRNA的表达
从筛选出的差异表达基因中挑选HMGCLL1、COA6基因进行荧光定量验证,结果显示400日龄高受精率公鸡睾丸中HMGCLL1 mRNA的表达是低受精率公鸡的2.86倍,且差异显著(P<0.05);并且高受精率公鸡睾丸中COA6 mRNA的表达量是低受精率公鸡的3倍,且差异显著(P<0.05)(图4)。
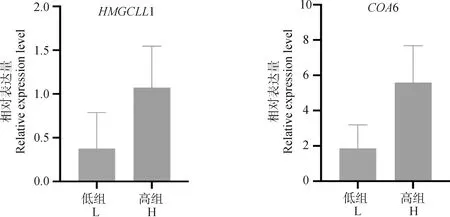
图4 HMGCLL1和COA6 mRNA在高、低受精率公鸡睾丸中表达
3 讨 论
过去十年里,GWAS使用高通量的基因分型技术在全基因组范围内寻找SNPs,探讨其与疾病以及复杂性状的关系[16]。在人类GWAS的研究中,已经鉴定出影响糖尿病、肥胖症、乳腺癌在内的几十种疾病的基因位点[17]。畜禽GWAS的研究中,研究人员也将GWAS应用在鸡[18-20]、猪[21-22]、牛[23-24]等重要经济性状上,与生长性能[25]、免疫[26]、产蛋[27]性能显著相关的SNPs位点和QTLs区域相继被挖掘,而对肉鸡孵化性状的GWAS分析研究较少。
白羽肉鸡在我国养禽业中占比高,其饲料转化率高、蛋白质含量高,是一种广受市场欢迎的禽肉类。生长、繁殖性状是肉鸡养殖业两大重要的经济性状,由于生长性能和繁殖性能选育方向不同,导致肉鸡在繁殖性状中的选育还有很大空间。本研究采用系谱和基因组信息估计了白羽肉鸡品系受精率性状遗传参数,受精率遗传力为0.14~0.21,属于低遗传力性状。本研究的ABLUP和GBLUP估计遗传力的结果与纯系蛋鸡报道的结果(受精率遗传力为0.16~0.33)基本一致。此外,利用全基因组关联分析筛选与白羽肉鸡受精率、孵化率相关的显著SNPs位点,利用转录组测序筛选白羽肉鸡受精率高、低组睾丸组织中的差异表达基因,利用生物信息学数据库分析差异表达基因参与的生物学过程,挖掘白羽肉鸡孵化性状相关基因,揭示孵化性状的遗传基础。全基因组关联分析结果表明,A群体556只个体的受精率筛选到4个显著基因,分别是位于12号染色体的COPG1基因、11号染色体的ADAMTS18、CDH8基因、4号染色体的FABP2基因;B群体398只个体受精率筛选到4个显著基因,分别是位于20号染色体的SLCO4A1基因、9号染色体的ATP13A3基因、1号染色体的NELL2、VEZT基因;B群体398只个体孵化率筛选到3个显著位点(P<4.57×10-8),分别位于18号染色体的TMEM、SCO1、MYH1A基因; A、B两群体芯片数据经plink软件合并后,954只个体受精率筛选到1个显著位点,位于5号染色体的IGHMBP2基因。转录组测序筛选到HMGCLL1、COA6等差异基因。4号染色体上的FABP2基因与受精率显著关联,有研究发现血浆中FABP2水平与血清胆固醇、尿酸、肌酐水平成正相关、并与白蛋白、红细胞和血红蛋白水平呈负相关,这表明FABP2水平随着糖尿病、肾病的严重程度而增加[28],该基因可能是影响肉鸡公鸡孵化性状的候选基因,同时鉴定出ADAMTS18、NELL2两个基因与受精率存在显著关联。研究发现,ADAMTS18参与小鼠生殖道的发育,2周龄小鼠敲除ADAMTS18后,雄性ADAMTS18/-小鼠的表皮腺整个腺体显著萎缩,7个月及以上的ADAMTS18/-小鼠的表皮腺体表面出现巨集观肿胀,最后得出结论ADAMTS18缺乏导致雄性小鼠的预发性腺发育不良和纤维化[29]。有研究发现,睾丸分泌蛋白(NELL2)通过ROS1途径发出信号,以调节附睾蛋白酶成熟和随后的附睾蛋白酶(OVCH2)分泌,该蛋白酶处理ADAM3以获得精子受精能力,最终得出结论,睾丸-附睾精子(NELL2-ROS1-OVCH2-ADAM3)信号传导是雄性生育力所必需的[30]。很多研究都证明以上基因和雄性生殖有关,但本研究筛选出的NELL2基因与受精率的关系鲜见报道,有待进一步的研究。转录组测序筛选出的差异基因中,HMGCLL1、COA6调控雄性受精率的机制尚不清楚,还需进一步研究。THYN1基因属于线粒体黄体酶家族,催化α氧化过程和β氧化过程的脱氢步骤,这些步骤与脂肪酸β氧化有关。其可缓解脂肪沉积和脂质代谢紊乱,从而显著改善家禽的产卵率,证明THYN1与家禽的繁殖性能有关[31]。COL6A3基因会影响蛋白精氨酸甲基转移酶7(Prmt7)在雄性生殖细胞中的表达,从而通过miRNA和目标基因调节精子增殖[32]。PARD3B基因与肥胖相关疾病有关[33],肥胖和糖尿病对雄性的精液品质有负面影响,并与睾丸激素水平低有关[34]。精液品质会影响到孵化性状[9],因此这些基因可能是影响孵化性状的候选基因。
4 结 论
本研究以白羽肉鸡为试验群体,基于系谱和基因型信息估计的受精率遗传力分别为0.21和0.14。使用GWAS和RNA-seq技术分析了与白羽肉鸡公鸡孵化性状相关的候选基因,GWAS筛选到12个SNPs位点与孵化性状显著相关,这些位点位于ADAMTS18、FABP2、NELL2等基因。RNA-seq技术鉴定到HMGCLL1、COA6、THYN1、COL6A3等差异表达基因,这些基因可能是影响白羽肉鸡孵化性状的候选基因。本试验结果为白羽肉鸡后续基因功能、品种改良等研究奠定了基础。
