锂硫电池中硒缺陷WSe2催化性能的理论研究
2023-02-25胡平澳张会茹
胡平澳, 张 琪, 张会茹
(合肥工业大学工业与装备技术研究院, 航空结构件成形制造与装备安徽省重点实验室, 合肥 230009)
锂硫电池具有高理论比容量(1675 mA·h/g)与能量密度(2600 W·h/kg)、 低成本和环境友好等优势[1~6], 是满足先进储能要求的新型二次电池系统. 然而, 锂硫电池仍然存在多硫化物在电解液中的溶解和穿梭、 硫及其放电产物的绝缘性、 在充放电过程中从硫转化到Li2S发生的体积膨胀以及锂负极表面的枝晶生长等问题, 导致其实际应用受到限制. 其中, 穿梭效应导致正极活性材料的损失, 严重影响锂硫电池的性能. 锂硫电池充放电过程中, 产生的长链多硫化物溶解在电解液中, 并在浓度差作用下向负极扩散. 这种多硫化物不断溶解穿梭以及沉积的现象会显著降低库伦效率和电池容量[7~10]. 为了解决上述“穿梭效应”带来的一系列问题, 改善锂硫电池的性能, 有效的途径是向正极材料中引入金属化合物催化材料(如金属的硫族化合物、 氧化物、 碳化物等), 通过对多硫化物的吸附和催化转化来有效解决穿梭效应带来的影响[11~16].
二维过渡金属硫族化合物具备高比表面积和有利于离子快速迁移的层状结构[17~19]. 其中, 二硒化钨(WSe2)具备良好导电性、 高体积能量密度和大层间间距(0.6500 nm), 有利于快速的离子传输和电化学性能的提升[20,21]. 基于此, WSe2作为锂硫电池正极和隔膜催化材料已实现了电池电化学性能的显著提升[22~24]. 研究表明, 二维过渡金属硫族化合物材料的活性主要来自边缘位点, 然而边缘位点数量有限, 使其催化性能的提升受到限制[25~27]. 为了进一步提高催化剂的性能, 近年来研究人员通过引入原子空位制造表面缺陷的策略, 使催化剂暴露更多的高活性位点, 同时调节催化材料的表面电子结构[28~32]. Zhang等[29]报道了一种由中空介孔碳球(HMC)和缺陷二硫化钼组成的新型复合宿主(MoS2-x/HMC)用于提高锂硫电池的性能. 富含硫空位的MoS2-x纳米片可以提供更多的活性位点, 以增强对多硫化物的吸附, 并提高其催化转化的能力. Sun等[33]设计了一种具有Se空位缺陷的VSe2-垂直石墨烯(VG)异质结负载在碳布(CC)作为锂硫电池的优质宿主. 结果表明, 缺陷赋予VSe2纳米片电化学诱导硫化的作用, 以促进多硫化物的吸附和转化, 从而使VSe2-VG@CC/S电极具有良好的循环稳定性.Wu等[34]采用理论计算方法发现硫缺陷提升了MoS2-x表面电荷密度, 为多硫化物转化提供了热力学和动力学驱动力, 最终促进了催化转化过程.
系统的理论计算能为揭示缺陷浓度与催化活性之间的关系提供理论依据[35~39]. 本文采用基于密度泛函理论计算, 考察了原始WSe2和不同浓度的Se空位缺陷(3.125%, 6.25%, 9.375%, 12.5%)WSe2表面上的多硫化物吸附能力、 锂离子迁移能力和多硫化物转化能力, 研究结果为硒化钨的表面缺陷改性策略提供了理论依据.
1 计算方法和模型
1.1 计算方法
所有计算均采用Materials Studio中的DMol3模块进行[40,41], 交换关联泛函使用的是基于广义梯度近似(Generalized gradient approximation, GGA)的Perdew-Wang 91(PW91)函数[42,43]. 全电子双数值加极化基组(Double numerical polarization, DNP) 被用来描述价电子的波函数[44]. 结构优化中能量、 力和位移的收敛性阈值参数分别为2.7212×10-4eV, 0.5442 eV/nm和5×10-4nm. 截断半径设为0.54 nm. 在内核处理中引入相对论效应, 核心电子处理采用Effective core potentials(ECP)[45]. 自洽场(Self-consistent field, SCF)的收敛范围设置为2.7212×10-5eV. 在表面优化部分, 使用5×5×1 Monkhorst-Pack布里渊区网格[46]. 所有多硫化物小分子均在2 nm×2 nm×2 nm的周期单元中进行优化, 表面结构优化设置真空层厚度为2 nm, 以避免周期性对计算带来的影响. 对于在不同空位缺陷浓度的WSe2表面上的锂离子迁移, 采用了线性同步转变(Linear synchronous transit, LST)/二次同步转变(Quadratic synchronous transit,QST)方法[47]进行过渡态搜索, 设置的最大的QST步数为10步. 锂硫电池充放电过程能量曲线按照如下步骤进行绘制[48]:

总反应式为
多硫化物分子(LiPSs)和材料表面之间吸附强弱根据吸附能计算, 可定义为
式中:Ead(eV)表示吸附能;ELiPSs@slab(eV)表示表面吸附LiPSs后体系的总能量;ELiPSs(eV)和Eslab(eV)分别表示LiPSs和表面的能量. 根据该定义, 吸附能Ead的绝对值越大, 表明LiPSs和表面之间吸附强度越高. 对于多硫化物在催化材料表面的吸附, 考察了多种可能的构型. 文中所示为相对能量最低的最优吸附构型.
1.2 表面模型
WSe2具有典型的二维过渡金属硫族化合物(TMDs)晶体结构, W原子层处于两层六方密堆积的Se原子之间形成Se-W-Se结构, WSe2原子层之间通过弱相互作用堆叠. 采用最大暴露面(001)面进行模拟计算, 构造包含48个原子的(3×3)超胞. 不同空位浓度的WSe2表面通过删除相应个数的表面Se原子获得. 如图1所示, 将无缺陷WSe2表面标记为WSe2(0), 删除1个、 2个、 3个或4个表面Se原子分别获得空位浓度为3.125%, 6.25%, 9.375%和12.5%的WSe2表面, 依次标记为WSe2(1), WSe2(2), WSe2(3)和WSe2(4). 对于空位位点的分布, 分别考察了直线型连续性空位分布[49]和之字形连续空位分布[50],其中前者能量较为优势, 故选择直线型连续性空位分布(具体信息见本文支持信息). 另外, 4种缺陷表面WSe2(1~4)的平均空位形成能分别为1.86, 1.93, 1.96和1.92 eV. 在WSe2高温退火条件下能够实现Se空位缺陷[51~53].

Fig.1 Top view of the WSe2 surface models with different vacancy concentrations
2 结果与讨论
2.1 多硫化物的吸附
锂硫电池中优异的催化材料应该具备适中的吸附能力, 即能够有效锚定多硫化物, 且不造成多硫化物裂解[54,55]. 研究了多硫化物S8, Li2S8, Li2S6, Li2S4, Li2S2和Li2S在完美WSe2(0)表面, 空位浓度为3.125%的WSe2(1)表面、 空位浓度为6.25%的WSe2(2)表面、 空位浓度为9.375%的WSe2(3)表面以及空位浓度为12.5%的WSe2(4)表面的吸附情况.
图2(A)~(F)分别为S8, 长中链多硫化物Li2S8, Li2S6和Li2S4及短链多硫化物Li2S2和Li2S在不同Se空位浓度的WSe2(X)(X=0~4)表面上的吸附构型与吸附能.可见, S8分子均未与5种WSe2表面成键, S原子与表面Se原子之间最短的距离分别为0.3692, 0.3761, 0.3668, 0.3728和0.3775 nm. 吸附物种S8@WSe2(0), S8@WSe2(1), S8@WSe2(2), S8@WSe2(3), S8@WSe2(4)对应的吸附能分别为-0.68, -0.53,-0.64, -0.62和-0.61 eV. 与完美WSe2(0)表面相比, 几种表面相应的吸附能略有降低但相差不大. 在放电过程中正极的S8分子首先与2个Li+和2个e-发生还原反应形成Li2S8, 在相应的Li2S8@WSe2(X)结构中, Li2S8分子均明显向表面靠近, 与表面Se原子形成Li—Se键, 成键距离分别为0.2798, 0.2749,0.2725, 0.2713和0.2719 nm. 对应的吸附能增加至-1.38, -1.33, -1.52, -1.47和-1.45 eV. 随后,Li2S8进一步与Li+以及e-反应形成Li2S6, Li2S6@WSe2(X)中Li原子与表面Se之间最短距离分别为0.2876, 0.2713, 0.2861, 0.2831和0.2952 nm. 相对于Li2S8吸附物种, Li-Se距离拉长, 对应的吸附能也表现出略微降低(-1.24, -1.26, -1.34, -1.32和-1.29 eV). 对于中链多硫化物Li2S4@WSe2(X), 小分子与表面的成键距离缩短, 吸附能相较于Li2S6物种呈现增加的趋势(-1.39, -1.26, -1.41, -1.38和-1.35 eV). 由此可知, 相比于S8, 5种催化表面对于长中链多硫化物的吸附均明显增强, 其中对于Li2S6和Li2S4的吸附稍弱于长链Li2S8. 由图2还可知, 对于短链多硫化物Li2S2, Li2S2@WSe2(X)中两个Li原子均与表面存在相互作用, 对应的吸附能显著增加(-1.59, -1.82, -1.87, -1.88和-1.91 eV); 对于产物Li2S, 两个Li原子与表面的作用进一步增强, 对应的吸附能进一步增加(-1.71, -1.97, -2.40, -3.20和-3.22 eV). 由此可知, 催化材料表面对于短链多硫化物的吸附明显强于长中链多硫化物.
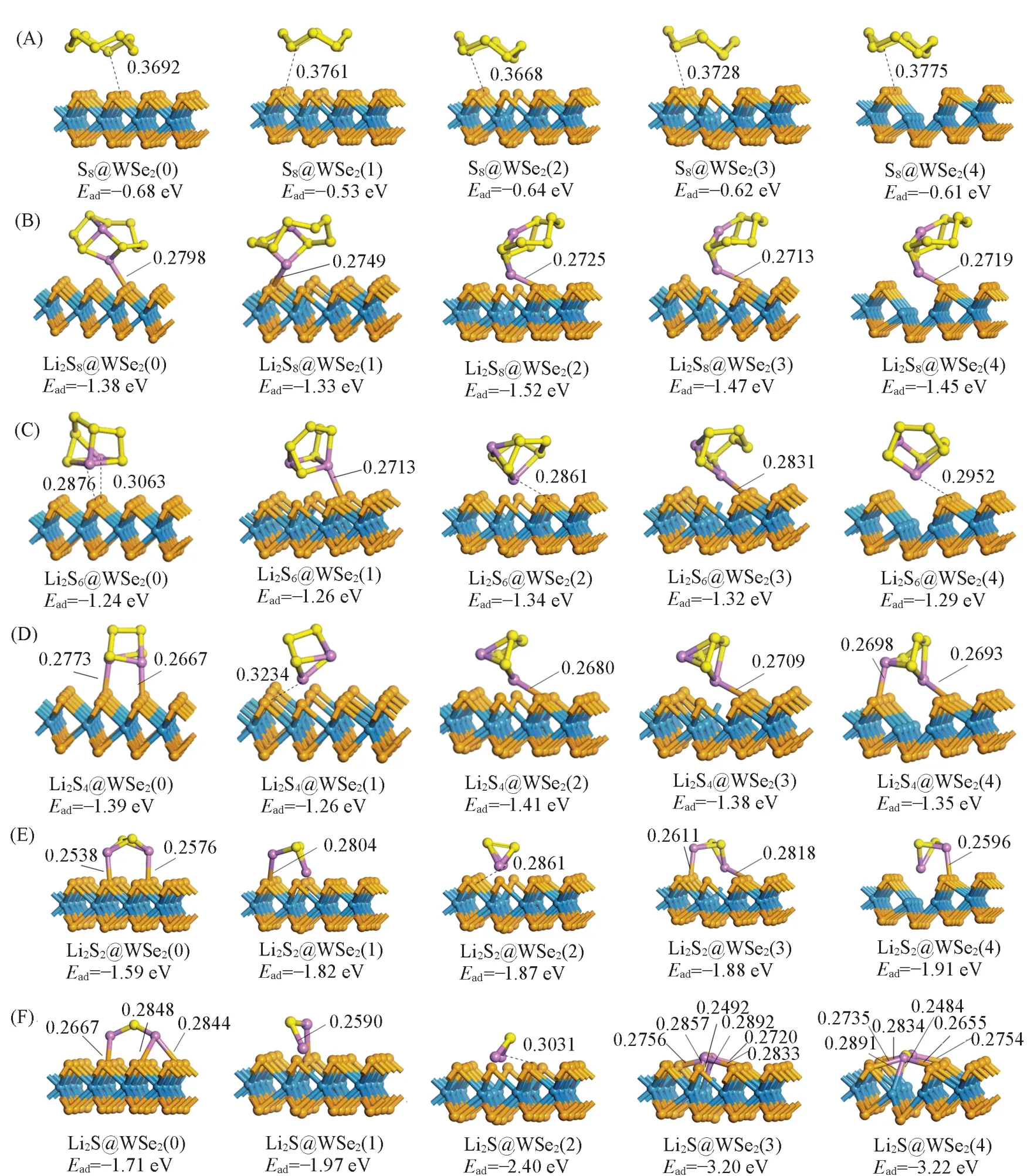
Fig.2 Stable adsorption configurations of S8(A), Li2S8(B), Li2S6(C), Li2S4(D), Li2S2(E) and Li2S(F) on five vacancy surfaces
为了进一步分析5种表面与多硫化物之间的相互作用, 将相应的吸附能示于图3中. 对于多硫化物之间的纵向对比和不同空位浓度WSe2表面之间横向对比得到如下规律: (1) 随着放电过程的进行, 吸附能力呈现先增强而后轻微降低, 最后显著增强的趋势. 即长中链多硫化物(Li2S8, Li2S6, Li2S4)的吸附明显强于S8物种, 但是Li2S6和Li2S4的吸附稍弱于长链Li2S8. 短链多硫化物(Li2S2, Li2S)的吸附明显强于长中链多硫化物. (2) 3.125%的低空位缺陷WSe2(1)表面不利于长链和中链多硫化物吸附, 其对于S8, Li2S8, Li2S4的吸附能低于WSe2表面.(3) 9.375%和12.5%的高空位浓度WSe2(3,4)表面对于长链多硫化物未呈现显著的吸附增强, 且对于短链多硫化物吸附过强. 如对于S8和Li2S4的吸附均弱于完美表面; 对于Li2S的吸附能高于3.0 eV.(4) 6.25%中等空位浓度的WSe2(2)对于长中链和短链多硫化锂均呈现适中的吸附能力增强.
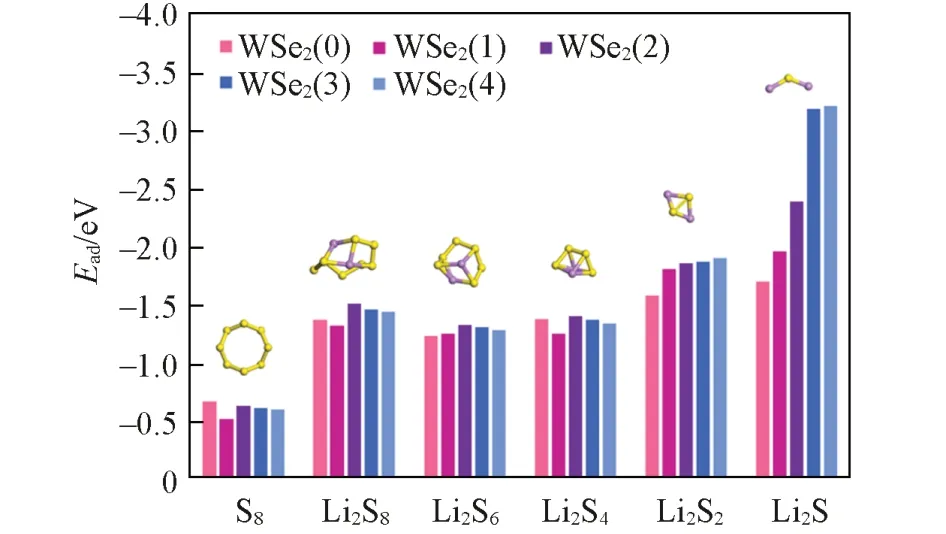
Fig.3 Comparison of the adsorption energies of S8 and Li2Sn(n=8, 6, 4, 2, 1) on five vacancy surfaces

Fig.4 Charge density difference of Li2S8 on the surface of WSe2(0)(A), WSe2(1)(B), WSe2(2)(C) and Li2S on the surface of WSe2(0)(D), WSe2(2)(E), WSe2(4)(F)
进一步通过差分电荷密度分析了不同空位浓度对于多硫化物的差异性吸附作用. 计算结果显示,低空位缺陷WSe2(1)表面对于长链多硫化物的吸附较弱. 基于此, 以Li2S8为例, 对比其在WSe2(0),WSe2(1)和WSe2(2)上吸附的差分电荷密度分布. WSe2(0)表面上失电子的青色区域集中在Li2S8, 得电子的黄色区域集中在表面, 吸附过程中Li2S8整体表现出向表面转移电荷的倾向. 对于WSe2(1)表面,黄色区域明显缩小, 且集中在多硫化锂与缺陷位之间. 说明Li2S8与表面空位之间存在相互作用, 但是Li2S8分子尺寸较大, 与单空位缺陷尺寸不匹配, 使得黄色区域较小. 相比之下, WSe2(2)表面与Li2S8之间黄色区域明显增加, 两者之间更为匹配的尺寸使得相互作用增强. 而这一尺寸匹配效应在短链多硫化锂中则不存在. 由于短链多硫化锂分子尺寸较小, 无论是低空位还是高空位缺陷, 均能与表面产生较强的相互作用. 如图4所示, Li2S与WSe2(0), WSe2(2), WSe2(4)表面之间均存在较大的电荷转移区域. 在WSe2(4)表面上, Li2S完全陷入缺陷空位中, 导致吸附作用过强, 不利于后续反应. 因此, 高空位浓度表面对于短链多硫化物表现出过强的吸附.
综上, 3.125%的低空位缺陷WSe2(1)表面由于尺寸匹配效应而不利于长链多硫化物的吸附;9.375%和12.5%的高空位浓度WSe2(3,4)表面对于长链多硫化物未呈现显著的吸附增强, 短链多硫化物由于陷入缺陷位点造成吸附过强. 相比之下, 6.25%中等空位浓度的WSe2(2)表面对于长链多硫化锂由于尺寸匹配而产生有效吸附, 对于短链多硫化锂则不会使其陷入空位, 因此对于所有多硫化锂均呈现适中的吸附, 在吸附方面的表现最为优异.
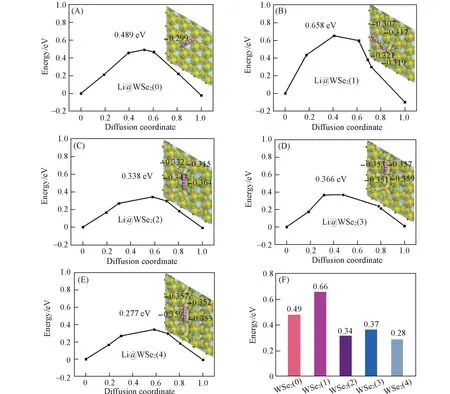
Fig.5 Energy profiles for Li diffusion on surfaces of WSe2(0)(A), WSe2(1)(B), WSe2(2)(C), WSe2(3)(D) and WSe2(4)(E), comparison of Li diffusion energy barriers on five vacancy surfaces(F)
2.2 锂离子的迁移
除了吸附作用之外, 材料表面锂离子迁移的快慢是衡量催化活性的另一重要因素[56~58]. 快速的锂离子扩散有助于硫物种的氧化还原反应. 通过计算锂迁移的能垒来研究不同空位浓度的WSe2表面锂离子的迁移. 图5所示为5种不同空位浓度WSe2表面的锂离子迁移路径和迁移能垒. 首先在表面选取稳定吸附位点, 确定迁移的初态和终态, 再通过过渡态搜索(TS-search)的方法得到迁移的能垒以及迁移路径. 在锂离子的迁移过程中, 所有表面均呈现先增大后减小的能量变化趋势.
图5(A)为WSe2(0)表面的锂离子迁移路径和迁移能垒, Li迁移的初始位点为W原子的hcp位, 迁移过程的终态为相邻W原子的hcp位. 在迁移过程中经过初态和终态的中点时达到能量最高点, 该过程的能垒为0.49 eV. 图5(B)为WSe2(1)表面的锂离子迁移路径和迁移能垒, Li从W原子的hcp位迁移至Se空位的hcp位过程能垒为0.66 eV, 逆过程能垒为0.78 eV.在Li迁移过程中经过Se原子附近时达到能量最高点. 单空位缺陷位点的存在, 使得锂离子迁移的能垒有所上升. 考察表面的Mulliken电荷分布, 发现WSe2(0)表面上Se原子电荷为-0.299 |e|, WSe2(1)表面上缺陷空位周围的Se原子电荷密度增加至-0.303~-0.321 |e|, 单空位缺陷使得Li与缺陷周围Se的作用更为紧密, 造成了表面上的一个能量势阱, 从而迁移能垒升高. WSe2(2), WSe2(3)和WSe2(4)表面的锂离子迁移路径相同, 其迁移路径和迁移能垒如图5(C)~(E)所示. 在这3种表面上, Li迁移的初始位点为Se空位的hcp位, 迁移过程的终态为相邻Se空位的hcp位. 在Li迁移过程中经过W原子的上方时达到能量最高点, 该过程的能垒分别为0.34, 0.37和0.28 eV. 均低于WSe2(0)和WSe2(1)表面上的迁移能垒. 这可能是由于直线型连续缺陷的产生, 使得Li迁移发生在相邻的缺陷位点之间. 尽管缺陷周围Se原子的电荷密度继续增加, 但是Li-Se之间的距离拉长至0.3154 nm[vs. WSe2(0)和WSe2(1)表面上的0.2494 和0.2483 nm], 使得迁移过程中Li-Se作用减弱, 从而降低迁移能垒.
图5(F)给出了5种不同Se空位浓度的WSe2表面上锂离子迁移能垒的对比. 发现随着空位浓度的增加, 迁移的能垒并不是单调变化的. 单个空位WSe2(1)表面的迁移能垒最高, 此时单个Se空位的存在阻碍了锂离子在Se空位周围的扩散. 除了单个空位的WSe2(1)表面提高了迁移能垒, 6.25%~12.5%的中高空位缺陷表面的锂离子迁移能垒都比原始WSe2表面低, 锂离子扩散能垒的顺序为WSe2(4)<WSe2(2)<WSe2(3). 因此, 3.125%的低空位缺陷表面不利于锂迁移; 6.25%~12.5%的中高空位缺陷表面可以促进锂离子的传输, 加速多硫化物的催化转化.
2.3 多硫化物的转化
为了研究不同空位浓度的WSe2表面对多硫化物的催化转化作用, 在不同电极电势(U)下, 对不同空位的活性位点上充放电过程的所有基元步骤开展进一步研究(图6), 通过过电势来评估不同空位浓度的WSe2表面的催化转化能力[48,59,60]. 充电电势(UC)定义为使得所有充电基元步骤刚好能够进行的最小电压, 放电电势(UDC)定义为使得所有放电基元步骤恰好不能进行的最大电压,U0为理论的热力学平衡电位. 基于此, 充电过电势(ηC)、 放电过电势(ηDC)和总过电势(ηTOT)的计算公式如下:ηC=UC-U0,ηDC=U0-UDC,ηTOT=ηC+ηDC. 图6(A)~(E)为WSe2(X)表面的充放电能量曲线、 以及对应的UC,UDC和U0,图6(F)汇总了5种表面的过电势ηC,ηDC和ηTOT.
在放电过程中, 正极活性物质S8经历S8→Li2S8→Li2S6→Li2S4→Li2S2→Li2S的过程得到放电产物Li2S. 如图6(A)所示, 在WSe2(0)表面, 正极活性物质S8从外电路和电解液分别获得2e-和2Li+得到长链多硫化物Li2S8, 能量下降至-5.78 eV(S8+2Li++2e-→Li2S8). Li2S8随后接受2/3e-和2/3Li+被进一步还原得到4/3Li2S6, 体系能量下降至-7.83 eV(Li2S8+2/3Li++2/3e-→4/3Li2S6). 之后, 4/3Li2S6接受4/3e-和4/3Li+被进一步还原得到2Li2S4, 体系能量下降至-11.82 eV(4/3Li2S6+4/3Li++4/3e-→2Li2S4). 2Li2S4接受4e-和4Li+被还原得到4Li2S2, 体系能量下降至-20.62 eV(2Li2S4+4Li++4e-→4Li2S2). 最后4Li2S2接受8e-和8Li+被还原得到8Li2S, 体系能量下降至-34.30 eV(4Li2S2+8Li++8e-→8Li2S). 上述放电过程的5个基元步骤的能量变化分别为-5.78, -2.05, -3.99, -8.80和-13.68 eV. 对应的电子转移分别为2e-, 2/3e-, 4/3e-, 4e-和8e-. 因此, 单位电子转移对应的能量变化为-2.89, -3.09, -2.99, -2.20和-1.71 eV. 基于此, 放电过程的电位决定步骤(PDS)为Li2S2→Li2S,UDC=1.71 V; 充电过程的PDS步骤为Li2S6→Li2S8,UC=3.09 V. 相应的ηDC和ηC分别为0.43和0.94 V. 类似的, 对于WSe2(1), WSe2(2),WSe2(3)和WSe2(4)表面, 放电过程的PDS步骤分别为Li2S2→Li2S, Li2S2→Li2S, Li2S4→Li2S2,Li2S4→Li2S2,UDC=1.86, 2.26, 2.51和2.54 V; 对应的ηDC为0.43, 0.23, 0.39和0.37 V, 不同催化表面上的放电过电势数值相差不大(跨度为0.20 V); 而充电过程的PDS步骤均为Li2S6→Li2S8,UC=3.20, 3.07, 3.13和3.08 V; 对应的ηC为0.91, 0.58, 0.23和0.17 V. 随着空位浓度的增加, 充电过电势呈明显的逐渐减小趋势, 数值跨度为0.74 V. 总过电势分别为1.34, 0.81, 0.62和0.54 V. 随着空位浓度的增加, 总过电势也呈减小的趋势. 因此, 总过电势主要受到充电过电势的影响, 均随着空位浓度的增加而减小, 6.25%~12.5%的中高空位浓度表面上多硫化物的转化反应较为优势.
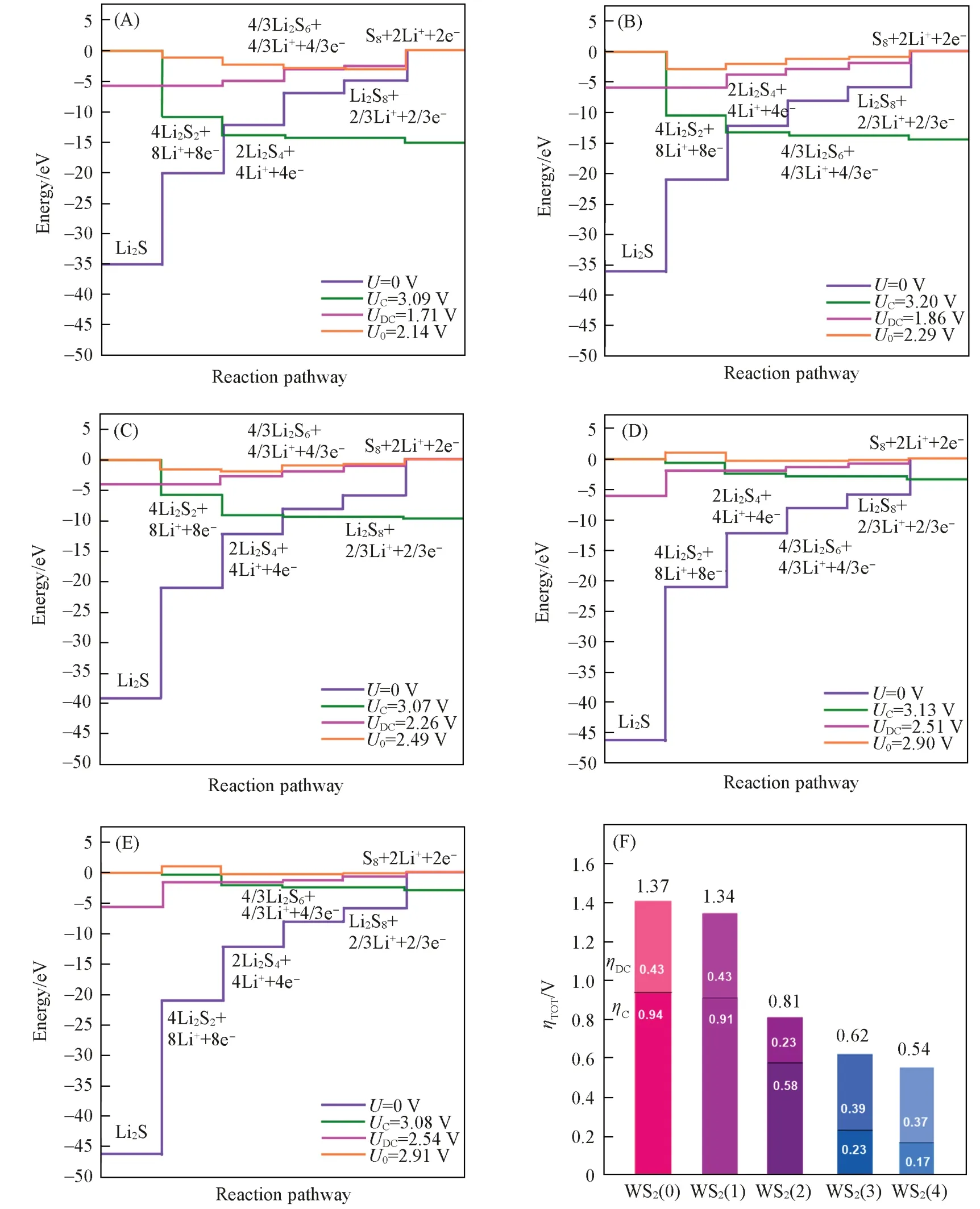
Fig.6 Free energy diagrams for charging-discharging process at zero potential(U=0 V), equilibrium(U0),charging(UC) and discharging(UDC) potentials on the five vacancy surfaces of WSe2(0)(A), WSe2(1)(B), WSe2(2)(C), WSe2(3)(D) and WSe2(4)(E), discharge(ηDC) and charging(ηC) overpotentials and their sum(ηTOT) for five vacancy surfaces(F)
基于上述计算结果, 3.125%的低空位缺陷WSe2(1)表面和完美表面WSe2(0)的充电放电过电势均相差不大, 表明低空位缺陷表面不能有效改善充放电性能. 9.375%和12.5%的WSe2(3, 4)表面的充电过电势明显低于WSe2(0)表面, 但是放电过电势与之相差不大, 未见显著降低. 相比之下, 6.25%中等空位浓度的WSe2(2)表面的充电和放电过电势均明显低于WSe2(0)表面, 即同时有利于充放电过程,是促进多硫化物转化过程的优势表面. 为了进一步阐述WSe2(2)表面的优异性, 以WSe2(0), WSe2(1)和WSe2(2)表面为例, 对比充电和放电过程的电位决定步骤. 如图7(A)所示, 三者的充电PDS均为Li2S6→Li2S8, 单位电子转移对应的能量变化分别为3.09, 3.20和3.07 eV, 单位电子转移所需能量越少, 则越有利于多硫化物转化. 该步骤能垒与Li2S6和Li2S8小分子在表面上的吸附紧密相关. 3种表面上Li2S8的吸附均大于Li2S6, 吸附能差值分别为0.14, 0.07和0.18 eV. 由于WSe2(1)对于长链Li2S8吸附较弱, 直接导致充电至Li2S8的过程较为困难; 而WSe2(2)对于Li2S8吸附增强, 该步骤相对容易. 3种表面上放电过程的PDS均为Li2S2→Li2S, 由图7(B)可见, 单位电子转移对应的能量变化分别为-1.71,-1.86和-2.26 eV, 单位电子转移所需能量越多, 则越有利于多硫化物转化. 3种表面上Li2S的吸附均大于Li2S2, 吸附能差值分别为0.12, 0.15和0.53 eV.由于WSe2(2)同样有利于短链Li2S的吸附, 因此放电至Li2S的过程相对容易. 由于6.25%中等空位浓度的WSe2(2)表面对长链和短链多硫化锂均具有优异的吸附能力, 而使其同时有利于充电和放电过程, 是促进多硫化物转化过程的优势表面.
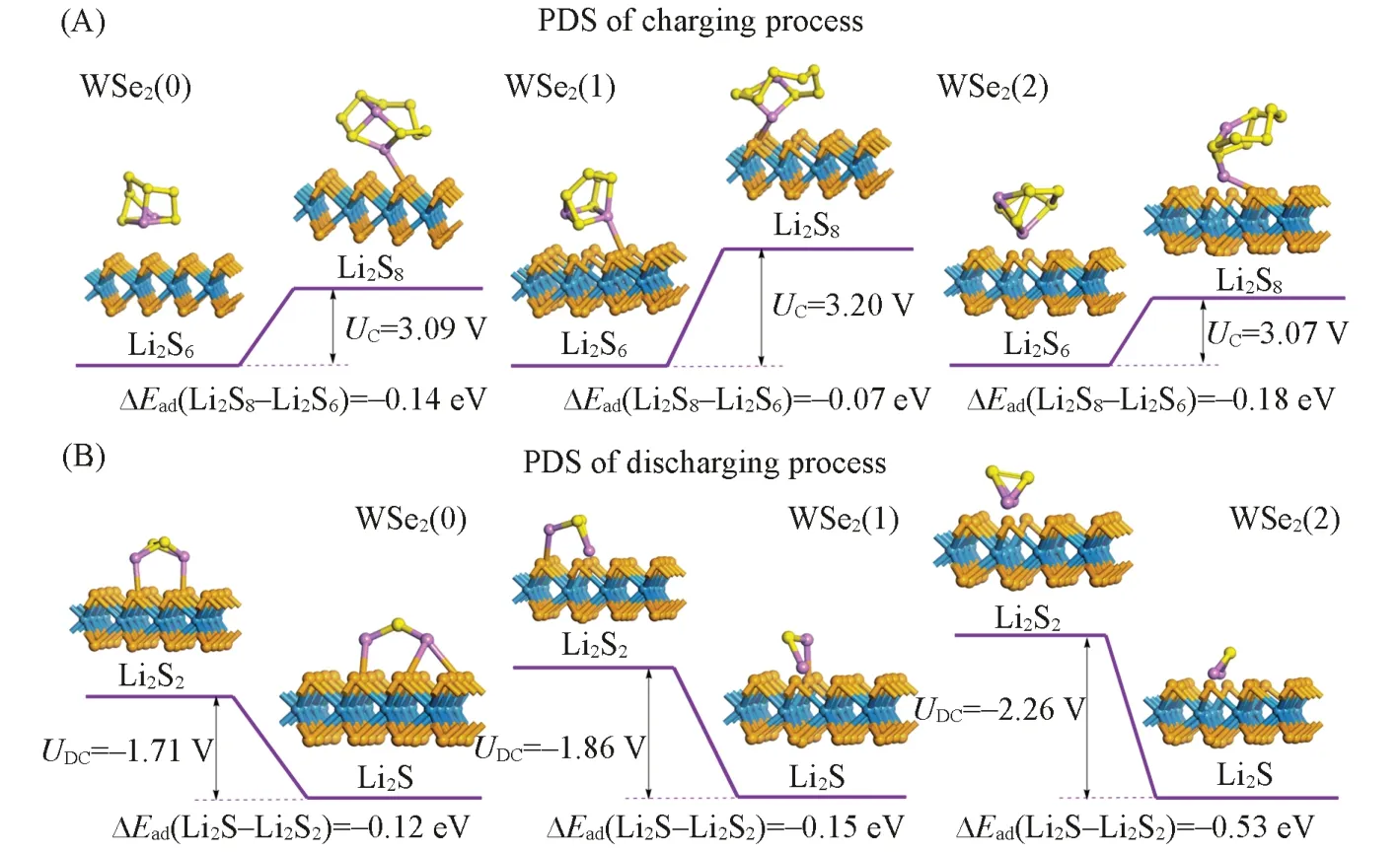
Fig.7 PDS of charging-process(A) and discharging-process(B) on the WSe2(0), WSe2(1) and WSe2(2) surfaces
2.4 空位浓度的影响机制
不同空位浓度的缺陷WSe2表面的催化作用体现在对于多硫化物的吸附、 锂离子迁移及多硫化物转化3个方面. 且随着空位浓度的不同, 三方面表现存在显著的差异.
对多硫化物吸附能力的考察结果显示, 3.125%的低空位缺陷WSe2(1)表面不利于长链多硫化物的吸附; 9.375%和12.5%的高空位浓度WSe2(3,4)表面对于长链多硫化物的吸附未见显著增强, 且对于短链多硫化物吸附过强. 相比之下, 6.25%空位浓度的WSe2(2)表面对于所有多硫化物均呈现适中的吸附能力增强. 基于锂离子迁移过程和能垒结果, 发现低空位缺陷WSe2(1)表面迁移能垒高于完美表面; 中高空位浓度WSe2(2~4)表面上, 迁移路径均为相邻Se空位的hcp位点之间, 迁移能垒均低于完美表面. 因此, 3.125%的低空位缺陷表面不利于锂迁移, 6.25%~12.5%的中高空位缺陷表面促进锂迁移.针对充放电能量曲线及过电势的考察, 3.125%的低空位缺陷WSe2(1)表面不能有效地促进多硫化物的转化, 9.375%和12.5%的高空位浓度表面不能有效促进放电过程. 6.25%中等空位浓度的WSe2(2)表面同时有利于充电和放电过程, 是促进多硫化物转化过程的最为优势的表面.
综合以上结果分析可得, 与原始表面WSe2(0)相比, 3.125%的低空位缺陷WSe2(1)表面的吸附相对较弱、 锂迁移能垒和充放电过电势较高; 9.375%~12.5%的空位浓度WSe2(3, 4)表面对于长链多硫化物吸附受限, 对于放电过程没有明显促进作用, 6.25%的空位缺陷WSe2(2)表面具有适中的吸附能力、 可观的锂离子传输能力和对于充放电过程的共同促进作用, 是最为优势的表面.
基于此, 锂硫电池催化材料性能主要体现在对于吸附-迁移-转化3个阶段的影响. 根据上述研究结果, 不同浓度缺陷对于长中短链多硫化物的吸附作用的差异性, 直接导致充放电反应过程中不同的电位决定步骤以及对应的极限电位. 基于此推测其它的二维催化材料(如MoS2, WS2等), 由于其本征结构、 电子性质不尽相同, 缺陷的产生对于吸附-迁移-转化3个方面的影响不同. 空位缺陷对于催化性能的影响可规律性地从吸附-迁移-转化3个方面考虑, 但是不同的体系产生的具体影响存在差异性.
3 结 论
对于完美WSe2表面和具有不同Se空位缺陷浓度(3.125%, 6.25%, 9.375%, 12.5%)的WSe2表面上的吸附、 迁移和催化转化进行了第一性原理研究. 结果表明, 3.125%的低空位缺陷WSe2表面在吸附方面不利于长链和中链多硫化物的吸附, 对于S8, Li2S8和Li2S4的吸附能低于WSe2表面; 在锂离子迁移中, 单个Se空位的存在阻碍了锂离子在Se空位周围的扩散, 呈现最高的迁移能垒; 在充放电过程中, 充电和放电过电势均与WSe2表面相差不大, 不能有效改善充放电性能. 9.375%和12.5%的高空位浓度WSe2表面对于长链多硫化物未呈现显著的吸附增强, 而对于短链多硫化物吸附过强; 虽然对于锂迁移和充电过程有促进作用, 但是放电过程的过电势与WSe2表面相差不大, 不能有效促进放电过程的动力学; 6.25%中等空位浓度的WSe2表面对于长中链和短链多硫化锂均呈现适中的吸附能力增强,对于锂迁移有明显促进作用, 且充电和放电过电势均明显低于WSe2表面, 即同时有利于充电和放电过程. 因此中等空位浓度使得WSe2表面具备适中的多硫化物吸附能力、 可观的锂离子传输能力和对于充放电过程的共同促进作用, 是多硫化物催化转化最为优势的表面. 该研究结果为缺陷改性硒化钨在锂硫电池中的应用提供了理论指导, 并希望能促进硒空位硒化钨在锂硫电池中的应用.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220595.
