气相色谱-质谱联用法测定液体调味料中4种氯丙醇的含量
2023-02-21刘沁颖毛燕妮卢跃鹏周晓婷
刘沁颖,侯 靖,毛燕妮,卢跃鹏,周晓婷
(1.武汉食品化妆品检验所,湖北 武汉 430040;2.国家市场监管重点实验室(食用油质量与安全),湖北 武汉 430040)
氯丙醇类化合物是一种人们公认的在食品加工过程中产生的污染物,包括3-氯-1,2-丙二醇(3-chloro-1,2-propanediol,3-MCPD)、2-氯-1,3-丙二醇(2-chloro-1,3-propanediol,2-MCPD)、1,3-二氯-2-丙醇(1,3-dichloro-2-propanol,1,3-DCP)和2,3-二氯-1-丙醇(2,3-dichloro-1-propanol,2,3-DCP),具有肾脏毒性、生殖毒性、免疫抑制和潜在致癌性[1-2]。1993年,世界卫生组织对氯丙醇类物质的毒性发出警告;2011年,国际癌症研究机构评估3-MCPD的毒性后将其归为2B类,认为它是一种对人类致癌性证据有限的可能致癌物。齐丽娟等[3]基于系统文献检索对3-MCPD及其酯类经口暴露的危害进行评估,动物研究显示3-MCPD毒性效应主要靶器官为肾和睾丸。动物长期低剂量暴露可引起雄性大鼠进行性肾毒性、睾丸毒性以及雌性大鼠肾毒性和乳腺增生,从而导致动物生育能力下降,表现出精子活性降低、产仔数量减少和完全不育等生殖毒性。小鼠短期高剂量暴露还可引起神经毒性作用。在最近的文献报道中,SEVIM Ç 等[4]通过大鼠脑组织中miR-21和PTEN-Akt等的表达情况,发现3-MCPD和缩水甘油亚可诱导大鼠脑组织凋亡。JIN C N等[5]采用3-MCPD灌胃大鼠28 d后,肾脏组织中发现有975个蛋白出现失调。生物信息学分析显示,3-MCPD可导致内源性代谢中与氨基酸、脂质和碳水化合物代谢相关的几种酶发生改变,参与这些通路的一些蛋白也发生了变化,这为肾毒性的分子机制提供了更全面的认识。LEE H S等[6]使用C2C12成肌细胞评估了3-MCPD的体外肌肉毒性,发现3-MCPD通过抑制mTOR和p70S6激酶表达从而对肌肉分化产生抑制作用。SCHULTRICH K等[7]研究发现,氧化应激可能在3-MCPD毒性中起作用,在肾脏、睾丸和大脑三个器官中,氧化应激的生物标志物不可逆氧化DJ-1蛋白的数量在喂食低剂量的3-MCPD时即显著增加。FAN Y等[8]研究发现,1,3-DCP作用于AKT/mTOR/FOXO1信号通路抑制自噬导致脂质积累,使实验小鼠表现出肝脏中甘油三酯、总胆固醇和脂滴数量增加。
液体调味料中氯丙醇的主要来源为水解植物蛋白,其为植物性蛋白质在酸催化作用下,水解后的产物,主要成分是氨基酸。由于植物性原料中有脂肪的存在,在使用过量盐酸水解时,甘油三酯也会水解为丙三醇,并与盐酸进一步反应产生各种氯丙醇。另外有报道[9]称纸制品生产过程中可能会添加增强抗湿性的环氧氯丙烷,当纸制品接触到液态食品时,其中的环氧氯丙烷可分解成氯丙醇,进而迁移到食品中造成污染。曾莹等[10]报道纸制品中1,3-DCP与3-MCPD迁移被普遍检出。因此,建立液体调味料中氯丙醇含量检测方法很有必要。
现有氯丙醇检测方法主要有气相色谱-质谱联用法(gas chromatograph-mass spectrometer,GC-MS)、毛细管电泳-电化学检测法[11]、高效液相色谱-荧光检测法[12]、近红外光谱法[13]、电化学传感器法[14]以及比色法[15]等,其中,使用最广泛的是气相色谱-质谱联用法。氯丙醇的提取分离多采用固相支持液液萃取技术,常用的载体有硅胶[16]、氧化铝[17-18]和硅藻土[19-21]。氯丙醇为极性化合物,为了提高检测灵敏度,需要进行衍生处理,常见的衍生剂有七氟丁酸酐[16-17,21]、七氟丁酰基咪唑[19]、六甲基二硅氮烷与三甲基硅基三氟甲烷磺酸盐[18]、环己酮[20]和苯硼酸[22-23]。近几年也有非衍生化检测氯丙醇的报道,但需要结合凝胶色谱[24]或高分辨质谱[25],难以推广。本研究拟参考美国分析化学家协会(association of official analytical chemists,AOAC)方法,选取酱油、鱼露及水解植物蛋白液为液体调味料的代表,采用商品化硅藻土柱进行固相支持液液萃取,七氟丁酰基咪唑衍生,气相色谱-质谱联用检测,通过对前处理条件进行一系列优化,建立了一种前处理简单快捷的液体调味料中氯丙醇含量测定方法,为分析液体调味料及其他食品中的氯丙醇提供参考。
1 材料与方法
1.1 材料与试剂
酿造酱油、鱼露、水解植物蛋白液:购于武汉市场。
正己烷、乙酸乙酯(均为色谱纯):德国Merck公司;氯化钠、无水硫酸钠(均为分析纯):国药试剂化学有限公司;七氟丁酰基咪唑(色谱纯):美国REGIS公司。
3-氯-1,2-丙二醇(3-MCPD)、2-氯-1,3-丙二醇(2-MCPD)、1,3-二氯-2-丙醇(1,3-DCP)、2,3-二氯-1-丙醇(2,3-DCP)、D5-3-氯-1,2-丙二醇(D5-3-MCPD)、D5-2-氯-1,3-丙二醇(D5-2-MCPD)、D5-1,3-二氯-2-丙醇(D5-1,3-DCP)和D5-2,3-二氯-1-丙醇(D5-2,3-DCP)标准物质(纯度均>99%):德国Dr.Ehrenstorfer公司。
1.2 仪器与设备
7890B-5977B气相色谱-质谱联用仪、DB-5MS色谱柱(30 m×0.25 mm×0.25 μm):美国Agilent公司;Vortex-Genie 2涡旋混匀器:美国Scientific Industries公司;N-EVAP111氮气浓缩仪:美国Organomation Associates Jnc.公司;大孔硅藻土柱(12 mL):北京迪马科技有限公司。
1.3 方法
1.3.1 试剂与标准溶液配制
配制20%氯化钠溶液、1 000 mg/L氯丙醇及氘代氯丙醇标准储备液、氯丙醇混合标准中间液(10 mg/L)及氘代氯丙醇混合标准工作液(10 mg/L)。
1.3.2 样品前处理
称取4 g液体样品于15 mL离心管中,对于不含食盐的试样,称样后加入1 g氯化钠,准确加入氘代氯丙醇混合标准工作液20 μL,超声混匀5 min,待净化。将液体全部转移至硅藻土小柱中,平衡10 min。以10 mL正己烷淋洗,弃去流出液,以15 mL乙酸乙酯洗脱氯丙醇,收集洗脱液于玻璃离心管中。在洗脱液中加入4 g无水硫酸钠,振摇,放置10 min后过滤,转移滤液至密闭性很好的透明具盖玻璃瓶中,氮吹浓缩至约0.5 mL(切忌浓缩至干),用2 mL正己烷溶解,涡旋混合。用气密针加入0.04 mL七氟丁酰基咪唑,立即拧紧瓶盖,涡旋混合30 s,于70 ℃反应20 min。取出冷却至室温,加入2 mL 20%氯化钠溶液,涡旋混合1 min,静置使水相和正己烷相分层,转移正己烷相,加入约0.3 g无水硫酸钠进行干燥后供气相色谱-质谱测定。系列标准工作液同步衍生化。
1.3.3 气相色谱-质谱分析条件
GC条件:DB-5MS毛细管柱(0.25 μm×0.25 μm,30 m);载气为高纯氦气(He)(纯度≥99.999%);流速为1 mL/min;进样口温度为250 ℃;不分流进样,进样量1 μL;升温程序为50 ℃保持1 min,以2 ℃/min升至90 ℃,再以40 ℃/min升至270 ℃,并保持5 min。
MS条件:电离方式为电子电离(electron ionization,EI)源;电子能量70 eV;离子源温度280 ℃;溶剂延迟时间8 min;监测方式为选择离子监测(selected ion monitoring,SIM),各化合物定量及定性、定量离子信息见表1。

表1 各化合物定量及定性离子Table 1 Quantitative and qualitative ion of each compounds
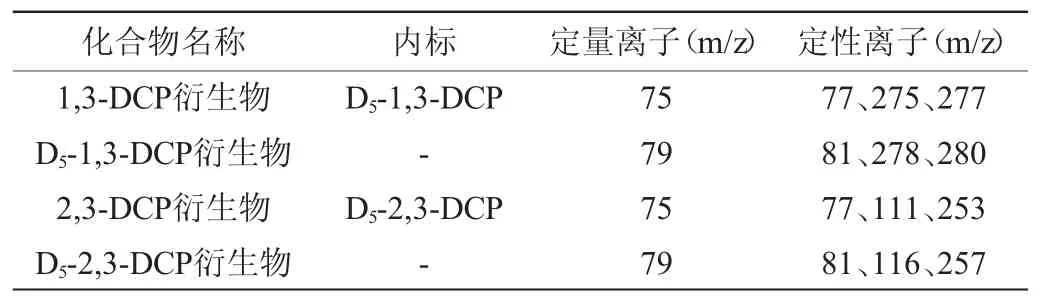
续表
1.3.4 数据分析
采用Agilent MassHunter 11.0软件采集数据并分析,采用Microsoft Office Excel 2016对实验数据进行图表绘制。
2 结果与分析
2.1 色谱柱的选择
氯丙醇的衍生物极性较小,一般使用弱极性色谱柱进行分离。因此,比较了5%苯基亚芳基聚合物毛细管色谱柱(DB-5MS)和5%苯基-甲基聚硅氧烷毛细管色谱柱(HP-5MS)对分离效果的影响。结果表明,同样升温条件下,使用HP-5MS色谱柱分离时,3-MCPD与2-MCPD无法完全分开,分离度为1.2;而使用DB-5MS色谱柱分离时,3-MCPD与2-MCPD可以基线分离,所以选择DB-5MS色谱柱进行实验。使用DB-5MS色谱柱对4种氯丙醇及其氘代内标物进行分离,总离子流色谱图见图1。

图1 4种氯丙醇及其氘代内标物GC-MS分析的总离子流色谱图Fig.1 Total ion chromatogram of 4 kinds of chloropropanols and their deuterated internal standards analyzed by GC-MS
2.2 淋洗溶液对结果的影响
正己烷淋洗可以除去提取液中的脂类等非极性干扰物,但是正己烷淋洗也可能会造成目标物的损失。分别称取4份液体样品4 g进行加标实验,分别用0、10 mL、20 mL和30 mL正己烷淋洗,检测淋洗液及后续15 mL乙酸乙酯洗脱液中氯丙醇含量。结果表明,不加正己烷淋洗不能有效除脂,后续实验过程中易出现乳化现象;1,3-DCP与2,3-DCP易被正己烷洗脱,10 mL正己烷淋洗即可使1,3-DCP与2,3-DCP发生明显的损失,3-MCPD与2-MCPD不易被正己烷洗脱;而20 mL和30 mL淋洗后,4种物质均有非常明显的损失。综合考虑,选择10 mL正己烷进行淋洗除脂。
2.3 洗脱体积对结果的影响
称取5份试样4 g液体样品进行加标实验,先使用10 mL正己烷淋洗,再分别用10 mL、15 mL、20 mL、25 mL和30 mL乙酸乙酯洗脱,检测洗脱液中氯丙醇含量。结果表明,4种氯丙醇的最佳洗脱体积均为25 mL,但1,3-DCP与2,3-DCP相比,3-MCPD与2-MCPD受洗脱体积的影响较小。在洗脱体积为15 mL时1,3-DCP与2,3-DCP洗脱含量约为最佳洗脱体积时的90%,3-MCPD与2-MCPD洗脱含量约为最佳洗脱体积时的80%。考虑到洗脱体积过大会显著增加后续氮吹的时间,增加检测成本,故洗脱体积确定为15 mL。
2.4 洗脱液吹干对结果的影响
氯丙醇为小分子化合物,沸点较低。为了考察溶液吹干对实验的影响,配备了含4种氯丙醇0.1 mg/L的溶液,一份在氮气下水浴吹干约5 min后拿出,2 mL正己烷复溶后衍生;另一份直接衍生。结果表明,1,3-DCP与2,3-DCP吹干后几乎全部损失,3-MCPD与2-MCPD吹干后也有较大损失,故洗脱溶剂切忌完全吹干。
2.5 衍生后溶液的稳定性
将衍生好的标准溶液分装在4个样品瓶中,分别在常温、避光常温、避光4 ℃和避光-18 ℃条件下保存,于0 d、1 d、2 d、3 d、5 d和7 d时上机检测(每次进样后立即更换新瓶盖,并移至规定条件下保存),记录各化合物在不同保存时间的峰面积。结果发现,4种氯丙醇及4种内标化合物在各条件下均非常稳定,7 d内没有明显的峰面积改变,说明衍生后的溶液稳定性良好。
2.6 方法验证
2.6.1 检出限、定量限与线性范围
分别称取4 g空白样品,加入一定量的氯丙醇标准中间液,制备含4种氯丙醇的加标样品,经处理后按1.3.3条件进行检测,以3倍信噪比计算检出限,以10倍信噪比计算定量限。以化合物质量浓度(x)为横坐标,峰面积(y)为纵坐标,得到回归方程,4种氯丙醇的检出限、线性范围与回归方程见表2。
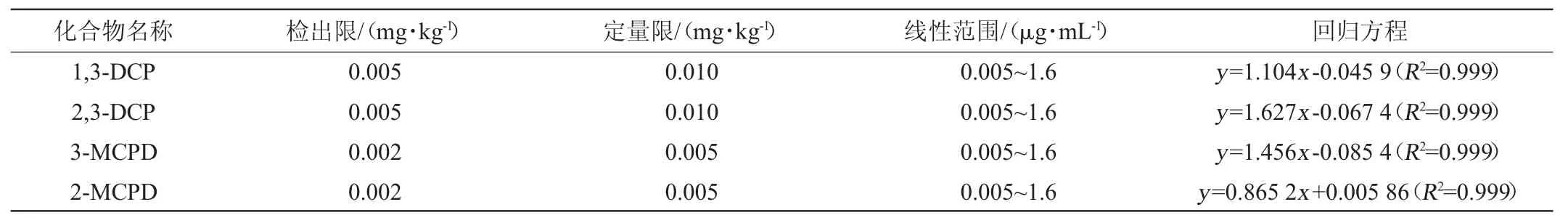
表2 4种氯丙醇的检出限、定量限、线性范围与回归方程Table 2 Limits of detection,limit of quantitation,linear ranges and regression equation of 4 kinds of chloropropanols
由表2可知,3-MCPD、2-MCPD、1,3-DCP和2,3-DCP的检出限分别为2.0 μg/kg、2.0 μg/kg、5.0 μg/kg、5.0 μg/kg,定量限分别为5.0 μg/kg、5.0 μg/kg、10 μg/kg、10 μg/kg,在0.005~1.6 μg/mL质量浓度范围内具有很好的线性关系。R2均为0.999。
2.6.2 回收率与精密度实验结果
分别称取4 g空白样品,分别加入氯丙醇,制备成加标样品。每个质量浓度的加标样品各6份,经处理后采用1.3.3条件进行检测,扣除未加标样品本底值后计算4种氯丙醇的回收率和精密度实验结果的相对标准偏差(relative standard deviation,RSD),结果见表3~表5。
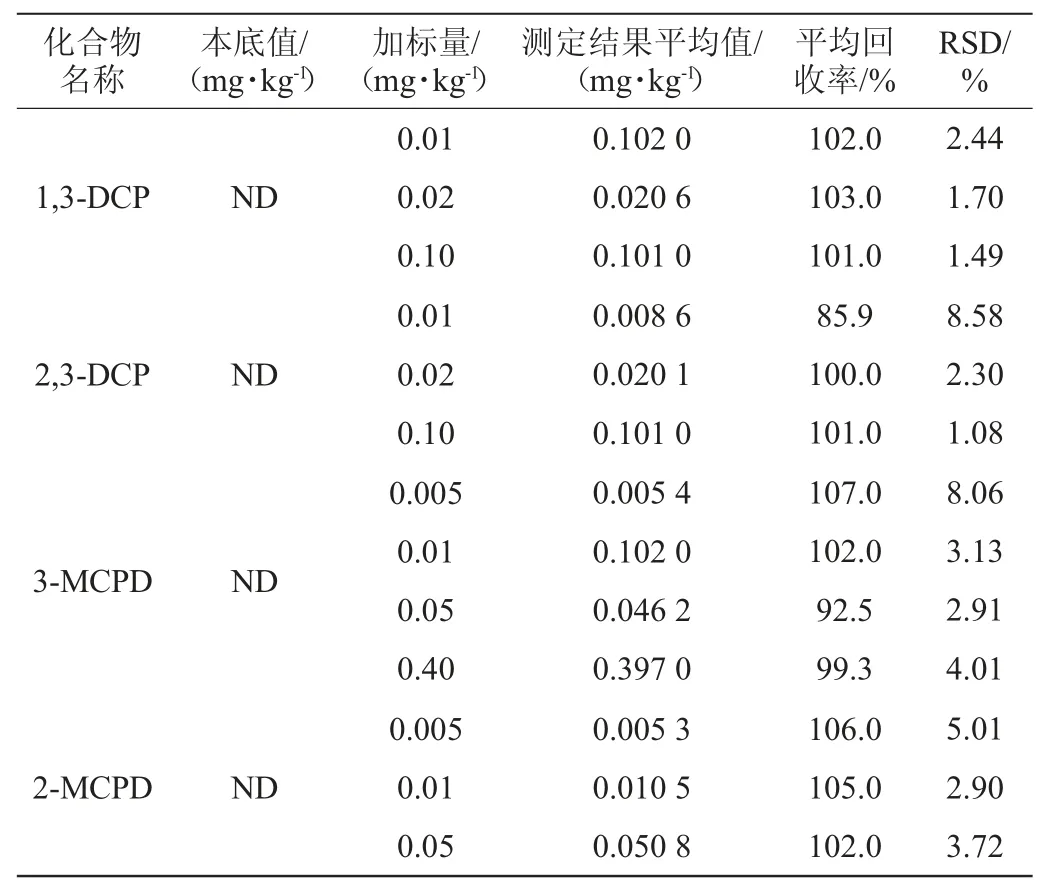
表3 酱油中4种氯丙醇回收率与精密度(n=6)Table 3 Recovery rates and precisions of 4 kinds of chloropropanols in soy sauce (n=6)

表4 鱼露中4种氯丙醇回收率与精密度(n=6)Table 4 Recovery rates and precisions of 4 kinds of chloropropanols in fish sauce (n=6)
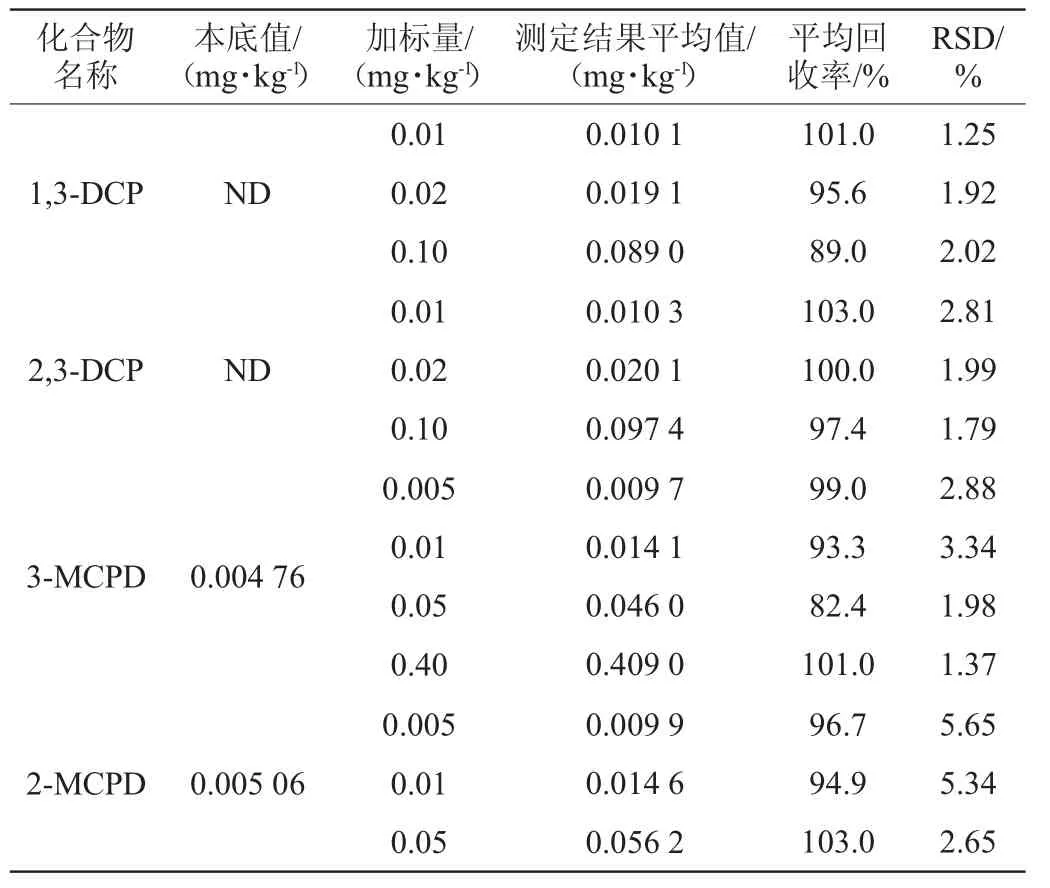
表5 水解植物蛋白液中4种氯丙醇回收率与精密度(n=6)Table 5 Recovery rates and precisions of 4 kinds of chloropropanols in hydrolyzed vegetable protein liquid (n=6)
由表3可知,酱油中4种氯丙醇的平均回收率在85.9%~107.0%范围内,RSD在1.08%~8.58%范围内;由表4可知,鱼露中4种氯丙醇的平均回收率在85.6%~106.0%范围内,RSD在0.85%~4.69%范围内;由表5可知,水解植物蛋白液中4种氯丙醇的平均回收率在82.4%~103.0%范围内,RSD在1.25%~5.65%。说明该方法应用于液体调味料中4种氯丙醇的检测时,回收率及相对标准偏差均在合理范围,方法的稳定性良好,是一种科学、有效、准确的检验方法,可以作为同类食品检测氯丙醇含量的参考。
2.7 实际样品测定
随机在市场购买酱油、鱼露、水解植物蛋白液各10份样品,经处理后采用1.3.3条件进行检测,结果表明所有样品均未检出氯丙醇。
3 结论
采用商品化大孔硅藻土柱净化,结合气相色谱-质谱联用法检测,建立了一种前处理简单快捷的液体调味料中氯丙醇含量测定方法。该方法测定液体调味料中3-MCPD、2-MCPD、1,3-DCP和2,3-DCP的检出限分别为2.0 μg/kg、2.0 μg/kg、5.0 μg/kg、5.0 μg/kg,定量限分别为5.0 μg/kg、5.0μg/kg、10 μg/kg、10 μg/kg。加标回收率为82.4%~107.0%,精密度实验结果的相对标准偏差为0.85%~8.58%。方法验证实验结果表明,该方法重复性好、准确度高,可以为市场监管和检验检测提供技术保障。
