UPLC-MS/MS法测定牛奶和豆乳中百草枯和敌草快的残留量
2023-02-21张立佳刘丽君高玉杰谢瑞龙吕志勇李翠枝
莫 楠,张立佳,刘丽君,高玉杰,谢瑞龙,吕志勇,李翠枝
(内蒙古伊利实业集团股份有限公司,内蒙古 呼和浩特 010110)
百草枯和敌草快均为联吡啶类季铵盐[1],是一种速效接触型灭生性除草剂,与土壤接触会迅速钝化,对根茎类作物无效[2]。百草枯和敌草快极易溶于水,在酸性溶液和中性溶液中稳定,在碱性溶液中易分解[3]。百草枯和敌草快具有很强毒性,其中尤以百草枯毒性最强,且尚无特效治疗方法[4-5]。百草枯和敌草快的广泛使用使其在动物性食品中再残留概率大大增加,我国标准GB 2763—2021《食品中农药最大残留限量》中规定,生乳中百草枯限量值为0.005 mg/kg,敌草快限量值为0.01 mg/kg。长期食用带有毒性残留物的食品对人体健康极易产生不良影响,因此,建立准确高效的检测方法尤为重要。
目前,对于百草枯和敌草快的检测方法主要有液相色谱法[6-8]、气相色谱-质谱法[9-10]、液相色谱-质谱联用法[11-12]、气相色谱法[13-14]。其中气相色谱法、气相色谱-质谱法检测最为普遍,但两种检测方法都存在百草枯沸点偏高而难以裂解和气化的缺点,同时前处理操作过于繁琐[15]。液相色谱法目前主要有反向色谱法和依托于亲水作用色谱柱法,这一方法优点在于可以实现碱性化合物的保留,同时又无需加入离子对试剂,但此方法也存在定性能力差、灵敏度偏低等缺点[16]。而液相色谱-质谱联用法具有选择性好、背景干扰少、灵敏度高等特点,作为确证方法最为合适[17-18]。
已报道的标准方法和文献中,牛奶中敌草快和百草枯单独测定或同时测定的极为少见。目前已报道的研究中多以粮谷、果蔬、水和生物样本为主,提取条件复杂且效率不高[19-21]。已有文献中,绝大多数采用纯有机试剂或酸化有机试剂来进行样品提取,这样的提取方法并不适用于牛奶和豆乳这类液体样品中百草枯和敌草快的提取,同时,净化条件也大多选用固相萃取小柱或固相填料进行样品净化,但这两种净化方式会对牛奶和豆乳中的目标物产生吸附作用,严重影响回收率[22-24]。本研究提出了先对样品进行酸化,使用甲醇沉淀蛋白,再加入正己烷除去脂类的方法,去除掉繁琐的净化方法,方便高效地改善了目标物的提取效率。本研究旨在开发一种简便、高效、准确测定牛奶和豆乳中百草枯和敌草快残留量的超高效液相色谱-串联质谱检测方法,对保障食品安全具有重要意义。
1 材料与方法
1.1 材料与试剂
敌草快二溴盐标准物质(C12H12Br2N2)(纯度≥99.0%)、百草枯二氯盐标准物质(C12H14Cl2N2)(纯度≥99.0%)、敌草快-d4(C12H8D4Br2N2)(纯度≥99.0%)、百草枯-d8(C12H6CL2N2D8)(纯度≥99.0%):德国Dr.Ehrenstorfer公司;Milli-Q超纯水、乙腈(质谱纯):德国Merck公司;甲醇(色谱纯):科隆化学品有限公司;甲酸(质谱纯):天津市沃尔孚科技发展有限公司;甲酸铵(色谱纯):上海安谱实验科技股份有限公司;WCX(3 mL,60 mg)固相萃取柱、HLB(3 mL,60 mg)固相萃取柱、C18(3 mL,60 mg)固相萃取柱、EMR-Lipid(3 mL,300 mg)固相萃取柱、PRIME HLB(3 mL,60 mg)固相萃取柱:美国Waters公司;牛奶、豆乳:市售。
1.2 仪器与设备
Thermo Biofuge Stratos低温高速离心机:美国Thermo公司;Waters TQ-S超高效液相色谱-串联质谱(ultra-performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)(配备电喷雾离子源):美国Waters公司。
1.3 方法
1.3.1 样品提取方式
方式①:称取5 g(精确到0.01 g)均匀试样(牛奶、豆乳),置于50 mL具塞离心管中,加入混合同位素标准工作溶液(1 μg/mL)75 μL,混合均匀,并加入50 μL甲酸混匀后超声20 min,涡旋震荡5 min。加入10 mL甲醇涡旋振荡5 min后,15 000 r/min条件下4 ℃离心10 min待净化。
方式②、方式③:分别按照参考文献[25-26]中的提取方法。
1.3.2 样品净化
将待净化液通过固相萃取小柱及正己烷净化(取1.2 mL上清液于塑料离心管中,加入0.3 mL正己烷,涡旋30 s,4 ℃、23 000 r/min离心10 min,吸取适量下层清液,0.22 μm微孔滤膜过滤),接取固相萃取小柱流出液,选择WCX、HLB、C18、EMR-Lipid、PRIME HLB柱以及正己烷进行净化试验,考察在不同的净化方式对目标产物平均回收率的影响。
1.3.3 色谱条件
Acquity UPLC BEH HILIC色谱柱(50 mm×2.1 mm,1.7 μm);柱温:35 ℃;进样量:2 μL;流动相:A为150 mmol/L甲酸铵溶液,B为乙腈;流速:300 μL/min;洗脱条件:0~3 min,40%A。
1.3.4 质谱条件
电离模式:电喷雾正离子模式;毛细管电压:0.9 kV;离子源温度:150 ℃;脱溶剂温度:500 ℃;脱溶剂气流量:1 000 L/Hr;碰撞室气流量:0.18 mL/min;多反应监测模式检测,其他质谱条件见表1。
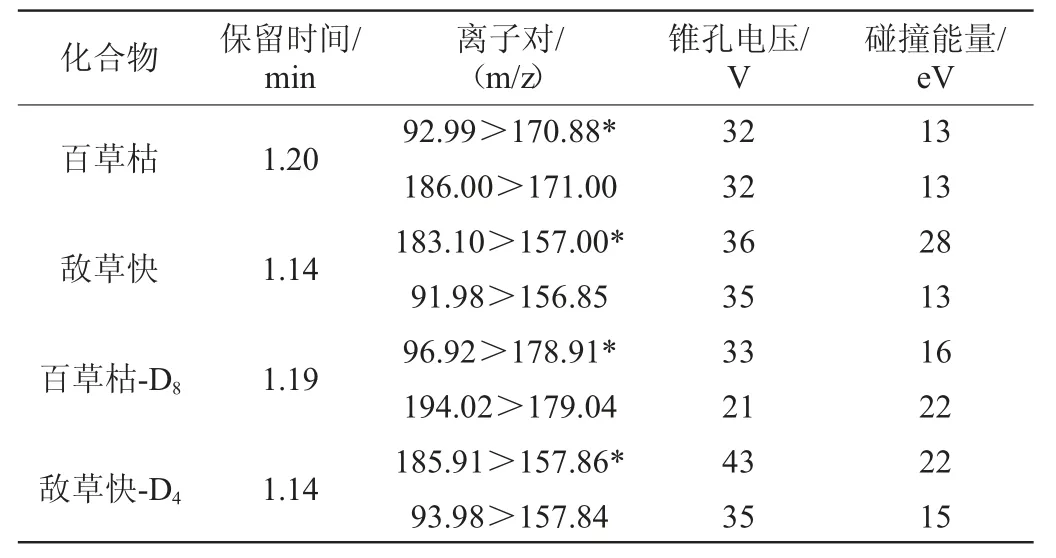
表1 百草枯和敌草快检测的质谱条件Table 1 Mass spectrum conditions for paraquat and diquat determination
1.3.5 标准曲线的绘制
按所需上机浓度准确吸取混合标准溶液和混合内标标准溶液,用空白基质溶液稀释,配制标准工作溶液,绘制标准工作曲线。
1.3.6 定性定量方法
定性:根据保留时间和离子的相对丰度定性。
定量:采用内标法定量。
1.3.7 基质效应分析
基质标准曲线的绘制:选择5份空白样品,采用本实验的前处理条件对样品进行提取、净化后,制备空白基质溶液。精密量取百草枯和敌草快混合标准工作溶液至不同的进样小瓶中,分别用空白基质溶液稀释,配制成质量浓度依次为0.5 ng/mL、1 ng/mL、2 ng/mL、5 ng/mL、10 ng/mL的系列标准工作液,采用UPLC-MS/MS测定,以响应值(Y)为纵坐标,以质量浓度(X)为横坐标绘制基质标准曲线。
溶剂标准曲线的绘制:精密量取百草枯和敌草快混合标准工作溶液至不同的进样小瓶中,分别用甲醇-1%甲酸水溶液(2∶1,V∶V)稀释,配制成质量浓度依次为0.5 ng/mL、1 ng/mL、2 ng/mL、5 ng/mL、10 ng/mL的系列标准工作液,采用UPLC-MS/MS测定,以响应值(y)为纵坐标,以质量浓度(x)为横坐标绘制溶剂标准曲线。
根据两种标准曲线,计算基质效应[27-28],其计算公式如下:

1.3.8 百草枯和敌草快含量的测定
待测样品中百草枯和敌草快的残留量按式(1)计算:

式中:X为待测样品中目标物残留量,mg/kg;C为待测样品中目标物上机质量浓度,ng/mL;V为样液定容体积,mL;n为稀释倍数;m为待测样品质量,g。
1.3.9 数据分析
使用waters MassLynx v4.1软件对数据进行数据采集,采用Origin 8.0绘图,采用Microsoft Excel 2016软件对数据进行处理。
2 结果与分析
2.1 样品提取方式选择
百草枯和敌草快极易溶于水,在酸性溶液和中性溶液中稳定,在碱性的溶液中不稳定,易分解。所以在前处理过程中选择在酸性条件下进行提取。依据国家标准[29],采用95%乙醇溶液进行提取,并不适用于牛奶和豆乳基质,净化程度和提取效率无法满足检测需求,故重新对提取溶液进行了选择。在相同称样量的条件下,分别使用该实验方式(方式①);甲醇-0.1 mol/L盐酸溶液(1∶9,V/V)[26](方式②)和甲酸-乙腈-水(5∶25∶70,V/V)[25](方式③)作为提取溶液进行相同浓度添加回收实验对比,比对结果采用外标法进行回收率计算,结果见图1。

图1 不同提取方式对2种农药回收率的影响Fig.1 Effects of different extraction methods on recovery rates of two kinds of pesticides
由图1可知,采用甲醇-0.1 mol/L盐酸溶液和甲酸-乙腈-水溶液提取百草枯回收率均低于30%。百草枯和敌草快以离子的形式存在于水中,在pH<7的溶液中比较稳定,同时酸性条件可以促进百草枯和敌草快的离子化,使其更容易由样品中迁移至水中,因此选择在称取样品中加入甲酸混匀后振荡并超声进行提取,促进目标物离子化并溶于水中[30]。后加入甲醇,进一步提取目标物和沉淀蛋白,避免因直接加入甲醇,而使样品蛋白沉淀过快,影响目标物的离子化而导致提取效率降低。本实验提取方法在保证回收率接近100%的提取效率的同时,又能够达到很好的沉淀蛋白的效果,适用于对牛奶和豆乳基质的提取处理。
2.2 样品净化条件选择
选择WCX、HLB、C18、EMR-Lipid、PRIME HLB柱以及正己烷进行同一浓度添加回收实验,采用本实验提取条件,上清液通过固相萃取小柱净化,接取固相萃取小柱流出液,过滤后上机,没有发现目标物,结果表明目标物已被固相萃取小柱吸附。后对固相萃取小柱进行洗脱,氮吹复溶上机,测定回收率,比对结果采用外标法进行回收率计算,结果见图2。
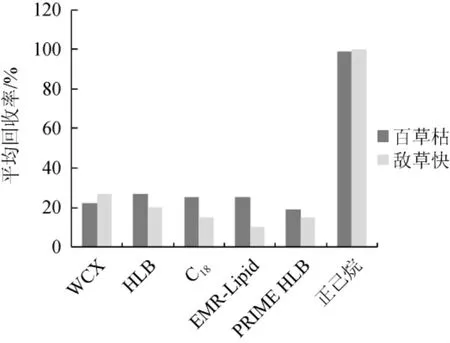
图2 不同净化方式对2种农药回收率的影响Fig.2 Effects of different purification methods on recovery rates of two kinds of pesticides
由图2可知,各固相萃取小柱填料均会对牛奶和豆乳基质中百草枯和敌草快产生吸附,且难以洗脱,绝对回收率均低于30%,正己烷处理条件下回收率高,故选择不使用固相萃取小柱净化,而采用正己烷除去脂肪的方式进行净化。此净化方式可有效的去除提取液中的弱极性物质,在保证目标物绝对回收率的同时,最大可能的去除提取液中的干扰物,方便经济高效。
2.3 流动相浓度的选择
由于百草枯和敌草快的强极性和正电荷,极易吸附在色谱柱上,并且难以洗脱,所以选用甲酸铵缓冲液作为流动相洗脱,甲酸铵可以起到加快出峰的同时增强目标物的仪器响应的作用,而这种影响与甲酸胺溶液浓度有着密切关系,故对甲酸铵的浓度进行优化。百草枯和敌草快均属于碱性化合物,需要酸性条件对峰型进行优化。以百草枯为例,选择在pH=3.7条件下,分别对10 mmol/L、50 mmol/L、100 mmol/L、120 mmol/L、150 mmol/L、160 mmol/L不同浓度甲酸铵缓冲液进行比对,结果见图3。由图3可知,在甲酸铵浓度为150 mmol/L和160 mmol/L时,可以得到较优的峰型,而甲酸铵浓度为160 mmol/L和为150 mmol/L时目标物峰型无明显变化,故最终选择150 mmol/L甲酸铵水溶液作为流动相。
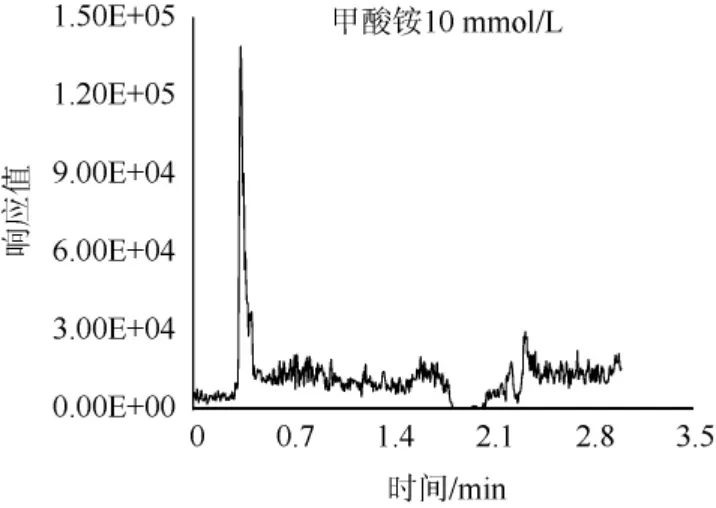
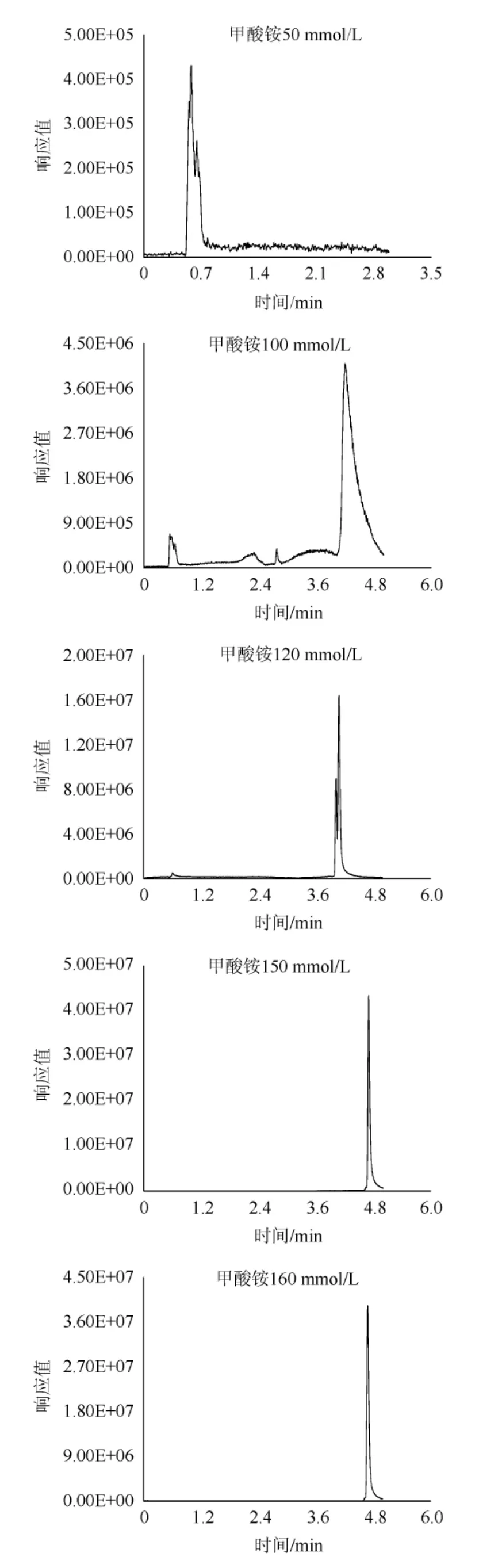
图3 不同浓度甲酸铵对百草枯出峰的影响Fig.3 Effect of different concentration of ammonium formate on the peak of paraquat
2.4 进样小瓶的选择
因为不同厂家的进样小瓶生产工艺存在差异,对目标物的影响也会不同,所以选择不同品牌的进样小瓶进行比对。使用空白基质溶液配制成相同浓度曲线标点,加入到两个不同厂家生产的玻璃进样小瓶中及塑料小瓶中,静置20 min后进行比对。结果表明,两种品牌玻璃进样小瓶中,敌草快和百草枯的浓度均有明显的下降。因为玻璃进样瓶内表面的自由离子溶解在样品溶液中,会暴露出玻璃表面活性位点,而百草枯和敌草快在溶液中均以离子化状态存在,会与玻璃表面活性位点吸附,导致目标物浓度降低,因此玻璃进样瓶并不适用于百草枯和敌草快的储存。又因为塑料进样小瓶不存在内表面暴露可与目标物结合的的活性位点的问题,不会导致目标物在样液的储存过程中产生损失,最终选择塑料材质进样瓶供上机检测使用。
2.5 色谱柱的选择
选取常用的UPLC BEH C18(50 mm×2.1 mm,1.7 μm)、UPLC HSS T3(50 mm×2.1 mm,1.7 μm)、UPLC BEH HILIC柱(50 mm×2.1 mm,1.7 μm)色谱柱进行比对,结果见图4。由图4可知,使用HILIC色谱柱的目标物峰型最为理想,因百草枯和敌草快极性强,在C18和T3色谱柱上难以保留,而目标物在HILIC色谱柱上可以很好的被保留,出峰时间适中,峰型和分离效果明显优于前两款色谱柱,因此,最终选定HILIC柱作为分离色谱柱。
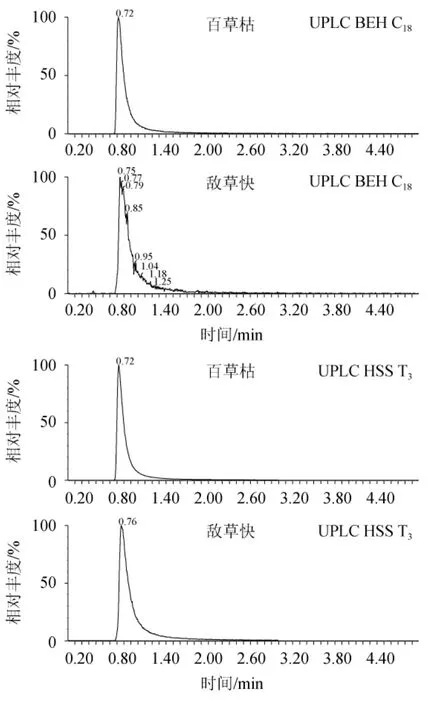
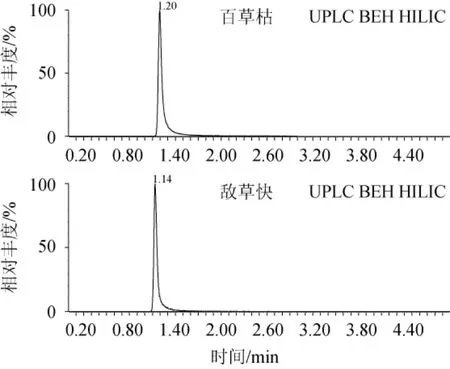
图4 不同色谱柱对百草枯和敌草快出峰的影响Fig.4 Effect of different chromatographic column on the peak of paraquat and diquat
2.6 基质效应实验
样品中含有多种物质,如乳糖、无机盐、磷脂等都可能会对目标物在离子源中的电离产生影响,无论是抑制还是增强目标物的电离,都会对定量目标物的准确性产生影响,所以需要对牛奶和豆乳的基质效应进行考察,以确保检测方法的准确性。针对不同基质样品,选取具有代表性的空白基质样品对基质效应进行了考查。用每种空白基质分别配制标准工作液并绘制工作曲线与前处理的提取溶液体系相近的甲醇-1%甲酸水溶液(2∶1,V/V)溶液配制的相同浓度点采用外标法所绘制的工作曲线相比较,基质抑制效应极其明显。后使用每种空白基质采用内标法分别配制标准工作液并绘制工作曲线与用甲醇-1%甲酸水溶液(2∶1,V/V)溶液配制的相同浓度点采用内标法所绘制的工作曲线相比较,二者曲线斜率做比值,比值大于100%的存在基质增强作用,小于100%的存在基质抑制作用,不利于目标物电离。牛奶和豆乳的基质效应试验结果见表2。
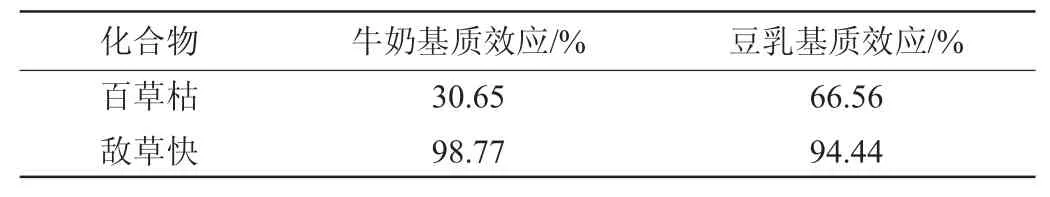
表2 各种样品的基质效应试验结果Table 2 Results of matrix effects of all samples
由表2可知,两种基质中百草枯有明显的基质抑制作用,而敌草快的影响得到了很好的削弱。推测这一现象是因为牛奶和豆乳空白基质对百草枯的外标和内标的离子化影响不一致导致。为避免基质效应对目标物定量的影响,在绘制标准工作曲线时,应选用内标定量和基质曲线相结合的方法进行配制标准工作液,以达到准确定量的目的。
2.7 标准曲线和检出限
依据优化后方法,对方法线性关系和检出限进行考察。用空白样品的提取液配制标准曲线,以响应值(Y)为纵坐标,以质量浓度(X)为横坐标绘制标准曲线,标准曲线及检出限见表3。

表3 百草枯及敌草快的标准曲线回归方程、线性范围、相关系数、检出限及定量限Table 3 Standard curve regression equation,linear range,correlation coefficient,limits of detection and limits of quantitation of paraquat and diquat
由表3可知,百草枯和敌草快在0.5~10 μg/mL质量浓度范围内,相关系数R均>0.99,线性关系良好。以目标峰等于3倍基线噪音为检出限,目标峰等于10倍基线噪音为定量限进行计算,百草枯和敌草快的检出限均为0.001 5 mg/kg,定量限均为0.002 5 mg/kg。
2.8 方法回收率和精密度
从每类样品中选取具有代表性的空白基质样品进行回收率试验,采用空白基质标准曲线,内标法定量,本研究百草枯和敌草快加标量分别为0.002 5 mg/kg、0.005 mg/kg、0.025 0 mg/kg,分别计算精密度试验结果相对标准偏差(relative standard deviation,RSD),结果见表4。
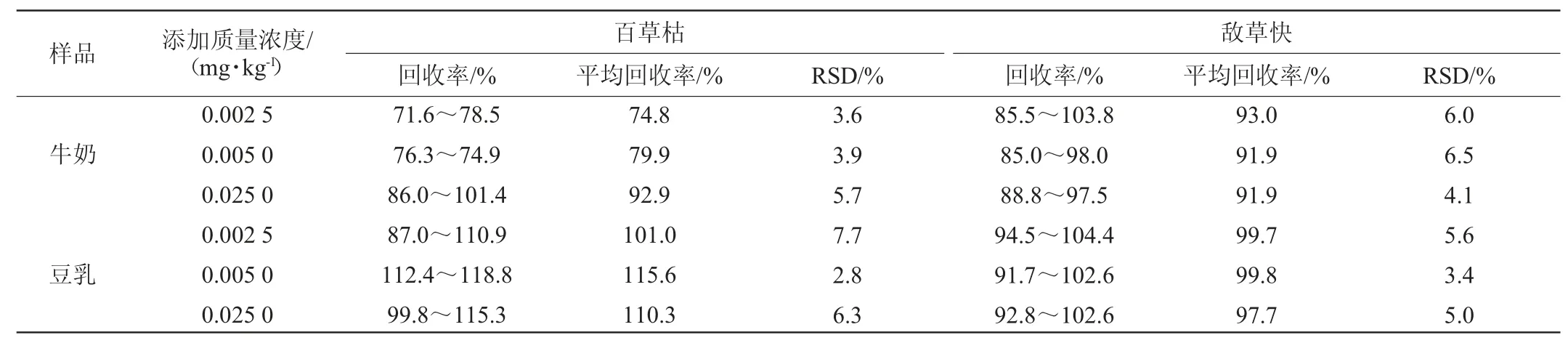
表4 方法回收率与精密度试验结果Table 4 Results of recovery rates and precision tests of the method
由表4可知,不同加标水平百草枯和敌草快的平均回收率范围在74.8%~115.6%之间;精密度试验结果相对标准偏差(RSD)的范围在2.8%~7.7%之间,两种基质测定结果的RSD均<10%,满足国标对检测方法回收率和精密度的要求,表明本方法具有良好的准确度和精密度。
3 结论
本研究建立了一种可同时提取牛奶和豆乳中百草枯和敌草快的方法。本方法快速高效,在样品中加入甲酸,使样品酸化,再加入甲醇沉淀蛋白,达到充分提取目标物的目的。当百草枯和敌草快的添加量为0.002 5~0.025 mg/kg时,其准确度和精密度均符合国标要求。在保证检测方法可靠性的同时,也为牛奶和豆乳中农药残留检测提供了一种新的思路。
