基于转录组测序解析南瓜子叶黄化的分子机理
2023-02-21熊兴伟王艺琴田怀志张素勤耿广东
熊兴伟,王艺琴,田怀志,张素勤,耿广东
(贵州大学 农学院,贵州 贵阳 550025)
蜜本南瓜属葫芦科南瓜属,是中国南瓜(Cucurbitamoschata)栽培种中品质较好的品种之一[1]。植株在苗期与快速生长期易发生自然黄化,影响植株的叶绿素(Chl)含量和光合效率,导致植株生长异常甚至死亡,造成作物减产[2]。自然界黄化突变体植株的发生主要受叶绿体和光合作用的共同影响,但具体的突变机制复杂,主要涉及叶绿体基因突变、Chl合成基因突变与激素失调等过程[3-4]。Chl主要分布在叶绿体类囊体膜中,是捕光复合体的主要组成部分,在光合作用中起着重要作用[5]。从谷氨酰胺-tRNA(Glu-tRNA)开始到Chl a和Chl b的合成包括15个步骤,涉及27个基因和15种酶。其生物合成与叶片颜色形成、叶绿体发育过程同时发生,每一步酶基因突变都会影响Chl的合成,导致植株颜色改变[6-7]。目前,在许多植物中发现基因突变而导致植株颜色变化的现象,如拟南芥[8]、芝麻[9]、水稻[10]、红掌[11]、黄瓜[12]、烟草[13]等。叶片突变有利也有害,如水稻、小麦等作物的叶色突变影响着种子的成苗率[14-15];而烟草、茶叶或花卉等叶色突变,通常可以创造性状优良的品种,产生较好的经济效益[13,16-17]。
近年来转录组测序(RNA-seq)已广泛应用于生物学研究,它可从整体水平研究基因的结构和功能,研究某一生物现象或生物过程的分子机理,并可用于差异表达基因分析、基因突变检测、遗传图谱绘制等[18],是进行组学研究的重要手段。Lyu等[19]通过高通量转录组分析自然黄化突变的栾树,鉴定了9个参与Chl代谢和14个参与类胡萝卜素生物合成途径的候选基因。Jiang等[20]对兰花叶色突变体进行转录组测序,鉴定了与光系统相关的7个基因,其中6个基因上调表达,1个基因(psbA)下调表达;该研究表明,在一定培养条件下,可能产生影响光系统Ⅱ D1蛋白的叶色突变体。Wu等[21]研究黄叶银杏突变体发现,158_mature和gma-miR2118a-3p靶基因参与色素生物合成,151_mature、ptc-miR396e-3p和aly-miR156a-5p可能是叶色突变的关键调控因子。汪端华等[22]在中国南瓜中分离筛选到稳定遗传银叶突变体48a,可作为标记性状应用于育种。但南瓜自然黄化突变鲜见报道,在分子机理方面研究更少。南瓜幼苗颜色变异可作为研究Chl合成与代谢[23]、光合系统中基因功能及其调控机制的理想材料,也是典型的分子育种材料。因此,本课题组以南瓜黄化突变体为试材,利用RNA-seq技术,探索南瓜叶色黄化的分子机制,以期为基因挖掘和标记开发提供参考,为南瓜高效育种奠定理论依据。
1 材料与方法
1.1 植物材料
南瓜黄化苗(MB21M)为蜜本南瓜的自然突变株(图1),材料来源于贵阳金黔农业科技有限公司,通过实验室组织培养保存。突变株表型为全株黄色,在子叶平展期之前正常生长。在南瓜幼苗第一片真叶初展时,分别剪取黄化苗与正常绿苗(MB21CK)的子叶,立即用液氮速冻后放于-80 ℃保存备用。试验设置3次重复。MB21M黄化苗子叶取样后一周死亡,无法产生少量叶绿素以供光合作用,而对照绿苗正常生长。

A,MB21M黄化苗;B,MB21CK绿苗对照。
1.2 试验方法
1.2.1 RNA提取、cDNA文库构建和测序
使用TRIzol®试剂盒[天根生化科技(北京)有限公司]提取南瓜子叶样品的总RNA。用带有Oligo dT的磁珠富集有poly A尾巴的mRNA,用DNA探针杂交rRNA,DNA/RNA杂交链用RNaseH选择性消化,再用DNaseⅠ消化DNA探针,纯化获得mRNA。将mRNA片段化,用随机N6引物进行反转录形成双链cDNA,cDNA末端补平并在5′端磷酸化,3′端形成一个突出“A”的黏性末端,再连接一个突出“T”的鼓泡状接头。连接产物通过特异引物进行PCR扩增,PCR产物热变性成单链,再用一段桥式引物将单链cDNA环化得到单链环状cDNA文库,由华大基因股份有限公司(深圳)使用Illumina HiSeqTM 2500平台进行高通量测序。
1.2.2 差异表达基因(DEGs)筛选
使用华大SOAPnuke软件进行过滤。首先去除包含接头的reads(接头污染),接着去除未知碱基N含量大于5%的reads,再去除低质量的reads(质量值低于15的碱基占该reads总碱基数的比例大于20%的reads),得到clean reads;使用HISAT将clean reads比对到南瓜参考基因组(Cucurbita_maxima_v1.1)序列。根据FPKM(每千碱基转录物每百万次映射读取的片段数)方法分析基因表达水平。DEGs筛选条件为错误发现率(FDR)<0.05和| log2fold change |(|log2FC|)≥1。
1.2.3 DEGs功能注释与实时荧光定量PCR(qRT-PCR)验证
采用BLASTx将unigene序列与公开的蛋白质数据库进行比对,包括基因本体论(GO)、京都基因和基因组百科全书(KEGG)。对于转录因子(TF),使用Getorf检测unigene的开放阅读框(ORF),通过Hmmsearch将ORF比对到转录因子蛋白结构域,再根据PlantTFDB描述的转录因子家族特征对unigene进行功能鉴定。
利用Talent qRT-PCR PreMix(SYBR Green)荧光定量检测试剂盒(天根)对14个与Chl代谢、光合作用-天线蛋白等相关的关键基因进行qRT-PCR分析。用Primer 5.0软件设计引物(表1),引物由生工生物工程(上海)股份有限公司进行合成。用RNAprep Pure多糖多酚植物总RNA提取试剂盒(天根)从叶片中提取总RNA,然后反转录成cDNA。扩增反应在Gene9600实时PCR检测系统(Bioer)中进行,循环曲线如下:95 ℃ 3 min;95 ℃ 5 s;60 ℃ 15 s,循环40次;然后绘制熔解曲线以检查特异性扩增。qRT-PCR包括3个生物学重复和3次技术重复,以18SRNA为内参基因,用公式Ratio=2-ΔΔCT计算DEGs的相对表达量。

表1 qRT-PCR所用特异引物
1.2.4 DEGs的GO和KEGG富集分析
根据GO与KEGG注释结果,将DEGs进行功能与通路分析,同时使用R软件中的Phyper函数进行富集分析,计算P-value,然后对P-value进行FDR校正得到Q-value,将Q-value≤0.05视为显著富集。
2 结果与分析
2.1 转录组测序分析概况
为了解样本数据的可靠性,试验首先分析了每两样品之间所有基因表达量的皮尔森相关系数,发现MB21CKr2与MB21CKr3、MB21Mr1与MB21Mr3之间的皮尔森相关系数≥0.96(图2),表明试验重复间的相关性较高,试验稳定性较好。

r1、r2、r3分别表示3次重复。
经转录组测序分析,每个样品平均产出6.16 Gb数据;样品比对基因组的平均比对率为54.13%,比对基因集的平均比对率为60.31%;共检测到26 445个表达基因,其中已知基因26 045个,预测的新基因400个;共检测出14 664个新转录本,其中14 244个属于已知蛋白编码基因的新可变剪接亚型,420个属于新蛋白编码基因的转录本。
2.2 DEGs筛选与qRT-PCR结果
从南瓜黄化苗和对照绿苗子叶中分别鉴定了25 043和25 132个表达基因,其中1 313和1 402个基因分别在黄化苗和对照中特异表达(图3-A)。在黄化苗中共检测到12 687个DEGs,其中上调表达的基因6 444个,下调基因6 243个(图3-B)。
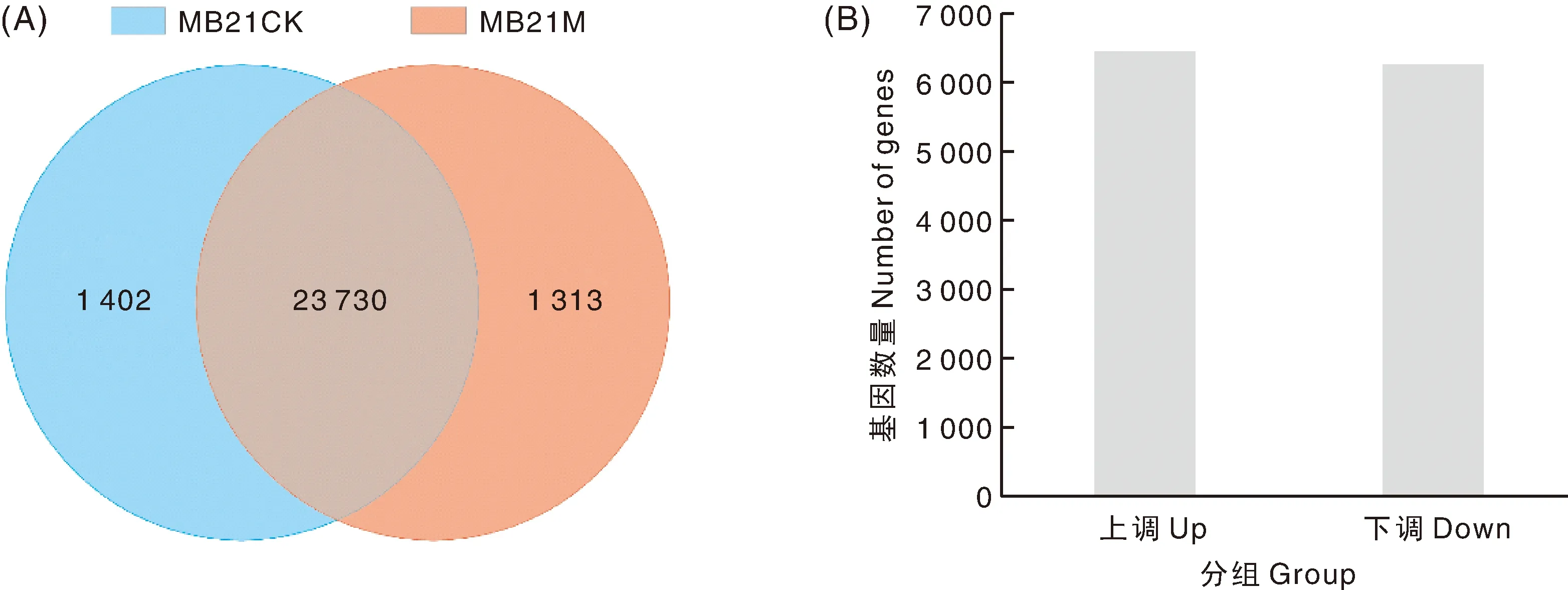
A,组间差异表达基因维恩图;B,突变体中差异表达基因数量。
为验证RNA-seq数据的可靠性,选择14个与光合色素代谢紧密相关的DEGs进行qRT-PCR分析。结果(表2)发现,RNA-seq和qRT-PCR相对表达水平之间的相关系数达到显著水平(r=0.833,P=2.15×10-4)(图4),证明RNA-seq数据可靠,转录组数据能准确反映南瓜黄化突变体基因表达水平的变化。
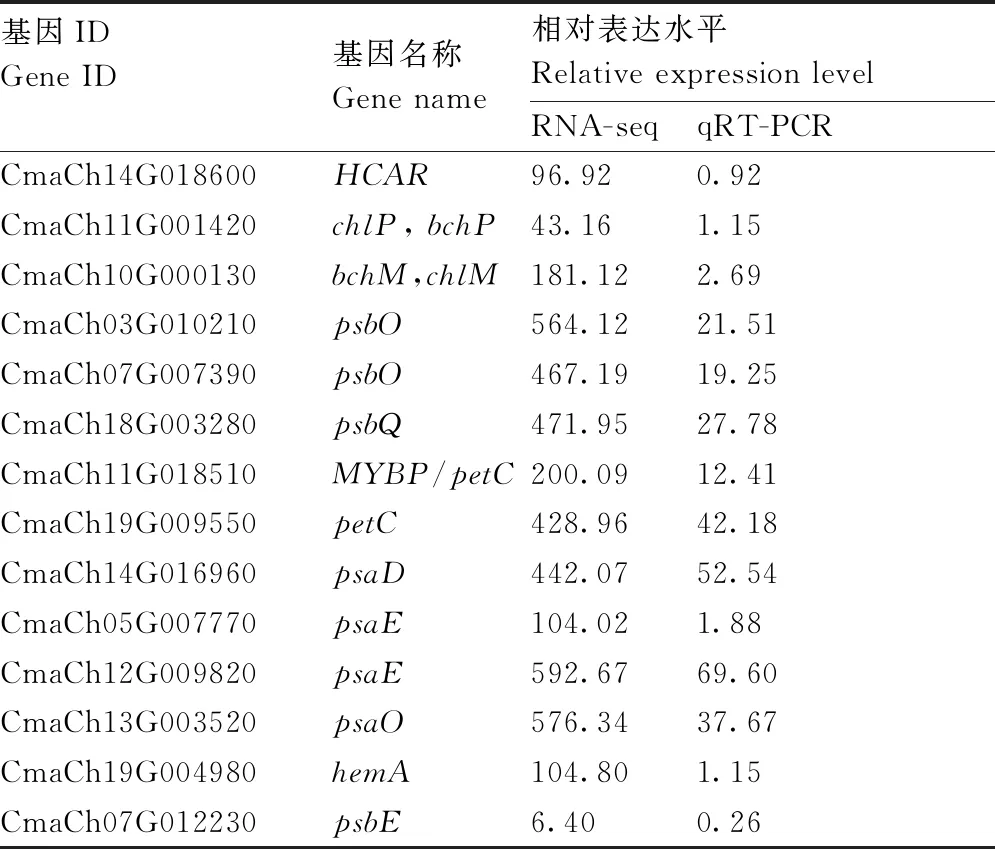
表2 qRT-PCR验证基因的名称与相对表达水平

图4 qRT-PCR和转录组数据的相关性
2.3 DEGs的功能注释与富集结果
GO分析表明(图5),7 311个差异基因主要富集到19个生物学过程、3个细胞组分和16个分子功能条目中。生物学过程中富集基因最多的是“细胞过程”和“代谢过程”。细胞组分中“细胞解剖实体”是富集最多的GO条目,依次是“细胞内”和“含蛋白质复合物”。分子功能中,差异基因主要参与“结合”与“催化活性”过程。GO富集气泡图分析表明,丝氨酸代谢富集比例较高(图6),在核苷酸结合过程中存在大量差异基因。另外,在分子功能类别中,筛选到与四吡咯结合序列相关的140个基因;在细胞组分中,有4 451个基因富集到细胞膜上,537个基因富集到细胞外周。说明这些富集途径对南瓜的叶色突变具有重要作用。
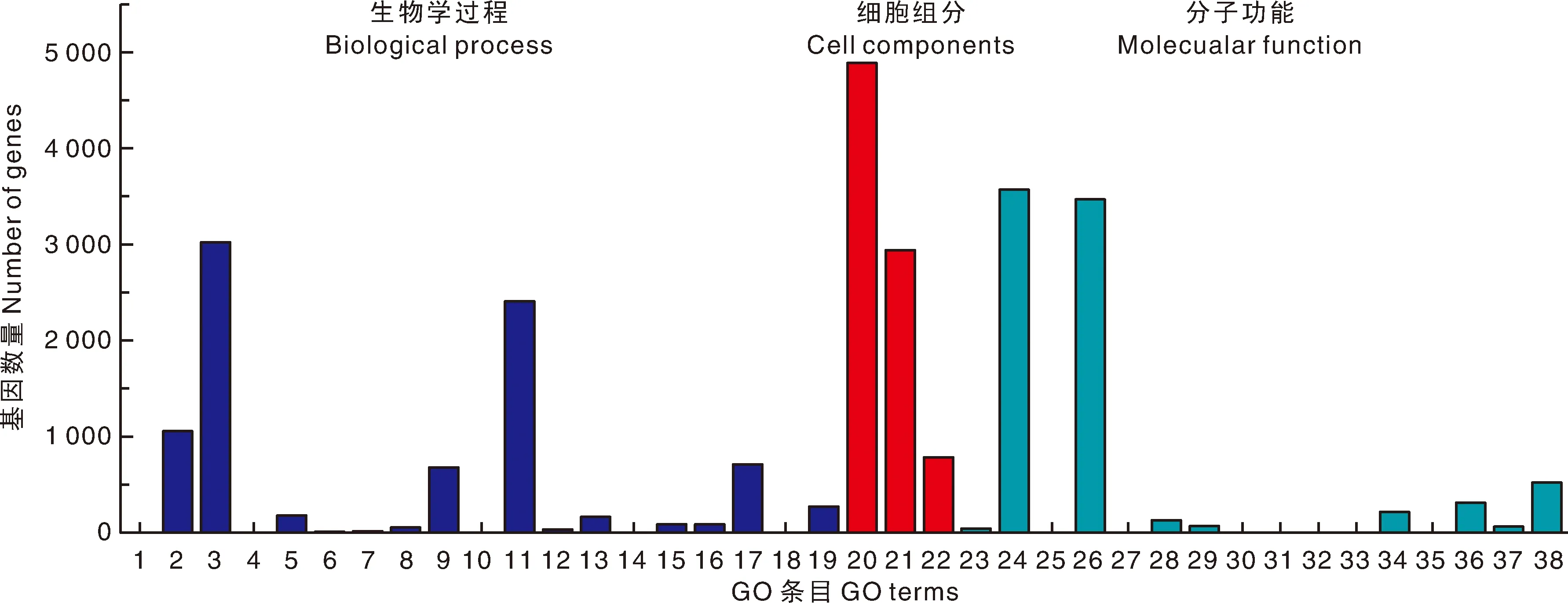
1,生物粘附;2,生物调节;3,细胞过程;4,排毒;5,发育过程;6,生长;7,免疫系统过程;8,物种间相互作用;9,定位;10,移动;11,代谢过程;12,多生物过程;13,多细胞组织过程;14,氮利用;15,繁殖;16,生殖过程;17,对刺激的反应;18,节律过程;19,信号转导;20,细胞解剖实体;21,细胞内;22,含蛋白质复合物;23,抗氧化剂活性;24,结合;25,受体活性;26,催化活性;27,分子载体活性;28,分子功能调节器;29,分子传感器活性;30,营养库活性;31,蛋白质折叠伴侣;32,蛋白质标签;33,小分子传感器活性;34,结构分子活性;35,毒素活性;36,转录调节活性;37,翻译调节活性;38,载体活性。
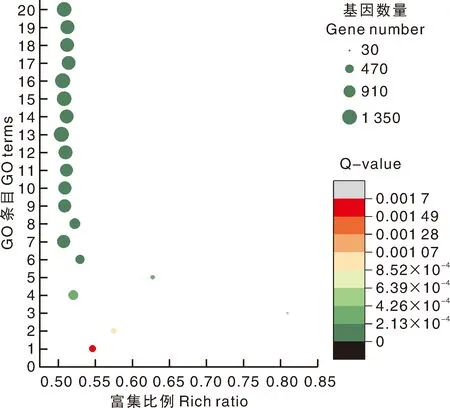
1,蛋白丝氨酸/苏氨酸激酶活性;2,细胞氨基酸代谢过程;3,丝氨酸家族氨基酸代谢过程;4,磷酸转移酶活性,醇基为受体;5,α-氨基酸代谢过程;6,蛋白激酶活性;7,催化活性,作用于蛋白质;8,激酶活性;9,腺苷酸结合;10,腺苷基核糖核苷酸结合;11,ATP结合;12,碳水化合物衍生物结合;13,阴离子结合;14,嘌呤核苷酸结合;15,核苷磷酸结合;16,小分子结合;17,嘌呤核糖核苷三磷酸结合;18,嘌呤核糖核苷酸结合;19,核糖核苷酸结合;20,核苷酸结合。
筛选了Q-value最小的前20个KEGG通路(图7),它们主要涉及MAPK信号通路-植物、内质网蛋白质加工、氨基酸生物合成等过程。富集因子最高的通路为光合作用-天线蛋白(photosynthesis-antenna proteins),富集因子为0.667。该通路中得到注释的基因有22个,全部是捕光Chla/b蛋白复合物(LHC)合成相关基因。富集到丙氨酸、天冬氨酸和谷氨酸代谢(alanine, aspartate and glutamate metabolism)的基因有65个,富集因子0.607;富集到卟啉和叶绿素代谢(porphyrin and chlorophyll metabolism)的基因有63个,富集因子0.583;硫代谢(sulfur metabolism)中有42个基因得到注释,富集因子0.609。光合作用-天线蛋白起着光能吸收和传递作用,卟啉和叶绿素代谢是合成叶绿素的通路,谷氨酸是叶绿素合成的前体物质,缺乏硫元素会使嫩叶发黄。由图可看出,这4个通路均有较高的富集因子和较小的Q-value值。
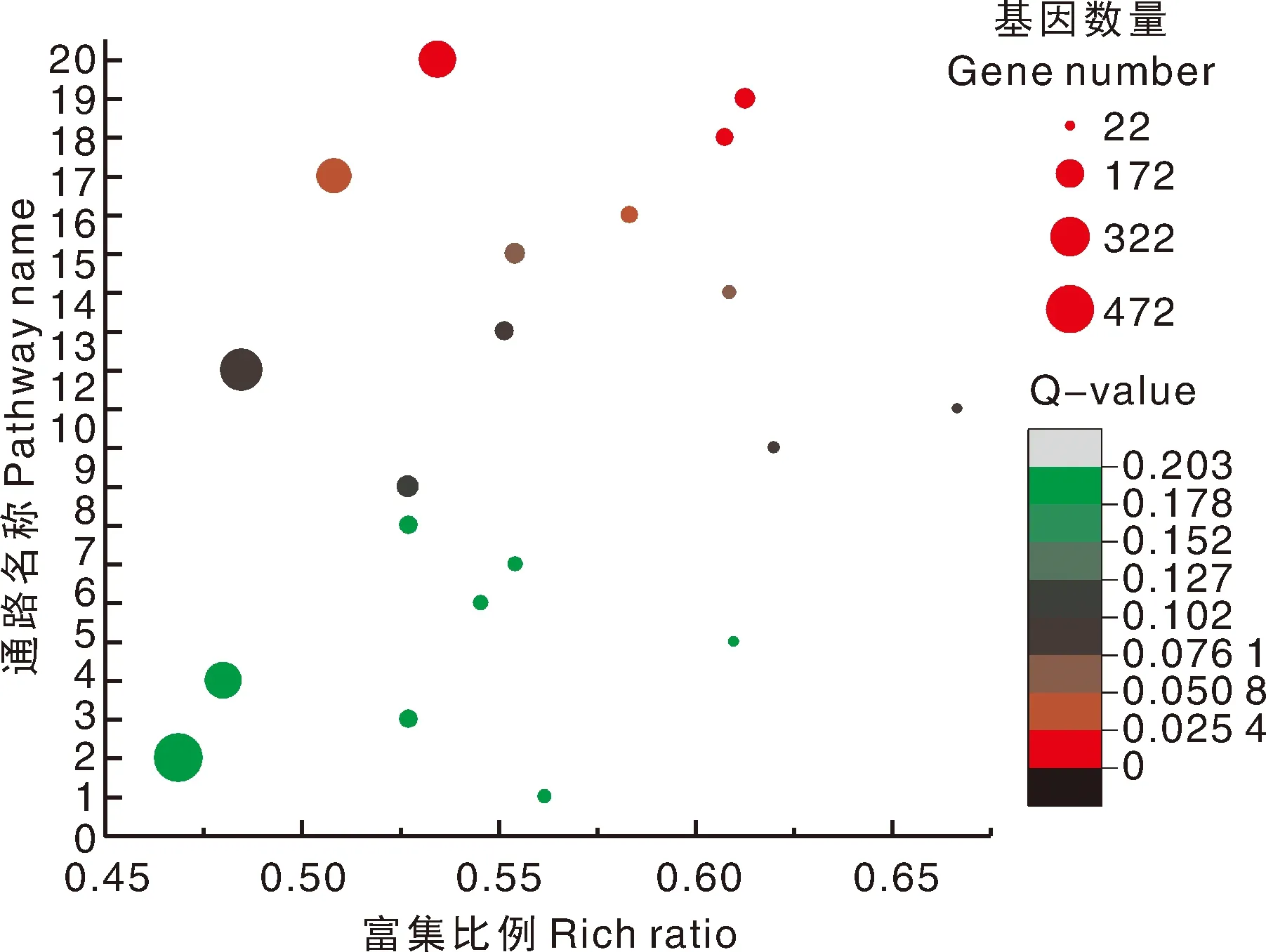
1,自噬-其他;2,植物激素信号转导;3,2-氧代羧酸代谢;4,碳代谢;5,硫胺素代谢;6,苯丙氨酸,酪氨酸和色氨酸生物合成;7,精氨酸生物合成;8,磷酸戊糖途径;9,过氧物酶体;10,丁酸代谢;11,光合作用-天线蛋白;12,MAPK信号通路-植物;13,甘氨酸,丝氨酸和苏氨酸的代谢;14,硫代谢;15,吞噬体;16,卟啉和叶绿素代谢;17,内质网中的蛋白质加工;18,丙氨酸、天冬氨酸和谷氨酸代谢;19,蛋白酶体;20,氨基酸生物合成。
对具有编码TF能力的基因进行预测,同时对基因所属的TF家族进行了分类统计。结果(图8)发现,2 321个DEGs被注释到58个TF家族,其中DEGs数量排名前5位的TF家族分别为MYB(301)、AP2-EREBP(242)、bHLH(212)、NAC(140)和WRKY(106)。
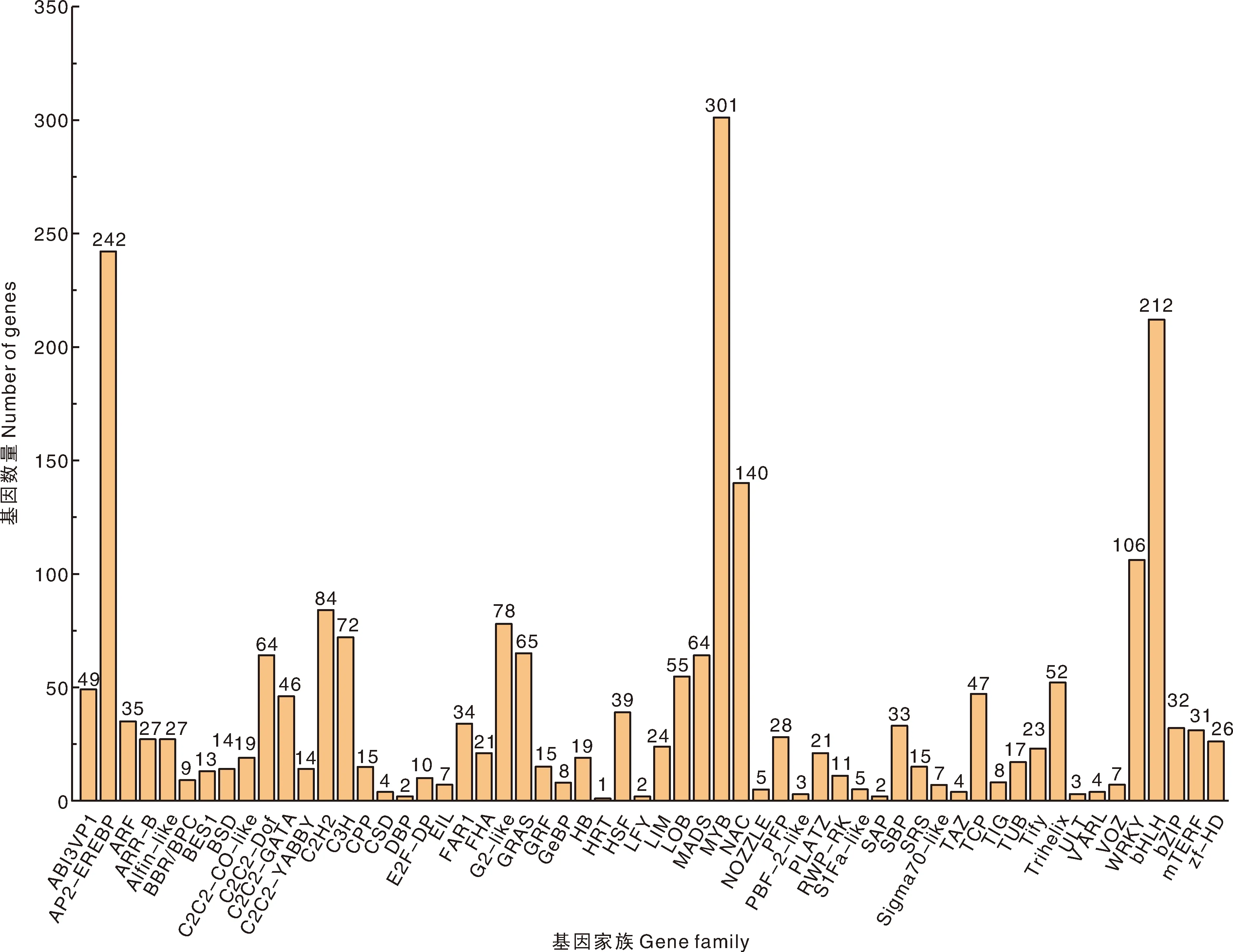
图8 DEGs所属转录因子家族分类
2.4 光合色素代谢相关DEGs的表达
根据卟啉与叶绿素代谢通路分析,以正常绿苗为参照,从黄化苗中检测到27个与Chl合成相关的DEGs(图9)。谷氨酰-tRNA还原酶基因(hemA)、谷氨酸-1-半醛-2,1-氨基转移酶基因(hemL)、粪卟啉原Ⅲ氧化酶基因(hemY)、8-乙烯基还原酶基因(DVR)、原叶绿素酸酯还原酶基因(Por)、7-羟甲基叶绿素还原酶基因(HCAR),编码镁螯合酶亚基Ⅰ(chlⅠ)、镁原卟啉O-甲基转移酶(chlM)、镁原卟啉Ⅸ单甲基酯环氧化酶(chlE)、叶绿素a合酶(chlG)等相关DEGs均下调表达。其中,hemA和hemL下调表达影响Chl合成初期从谷氨酸到5-氨基酮戊酸(ALA)合成过程[24]。hemY将原卟啉原Ⅸ转化为原卟啉Ⅸ,hemY的下调表达直接影响原卟啉Ⅸ的合成[25]。chlI催化Mg2+进入原卟啉Ⅸ生成Mg-原卟啉Ⅸ,chlM催化Mg-原卟啉原Ⅸ形成镁原卟啉Ⅸ甲脂(Mpe)[26],chlE催化Mpe生成二乙烯基还原酶[27],Por催化原叶绿素酸酯氧化形成叶绿素酸酯[28],chlG进一步催化合成Chl a[29],HCAR可协同叶绿素b还原酶将Chl b还原形成Chl a[30]。总之,该通路中绝大多数DEGs下调表达,抑制了Chl合成,其可能是导致黄化苗产生的重要原因。
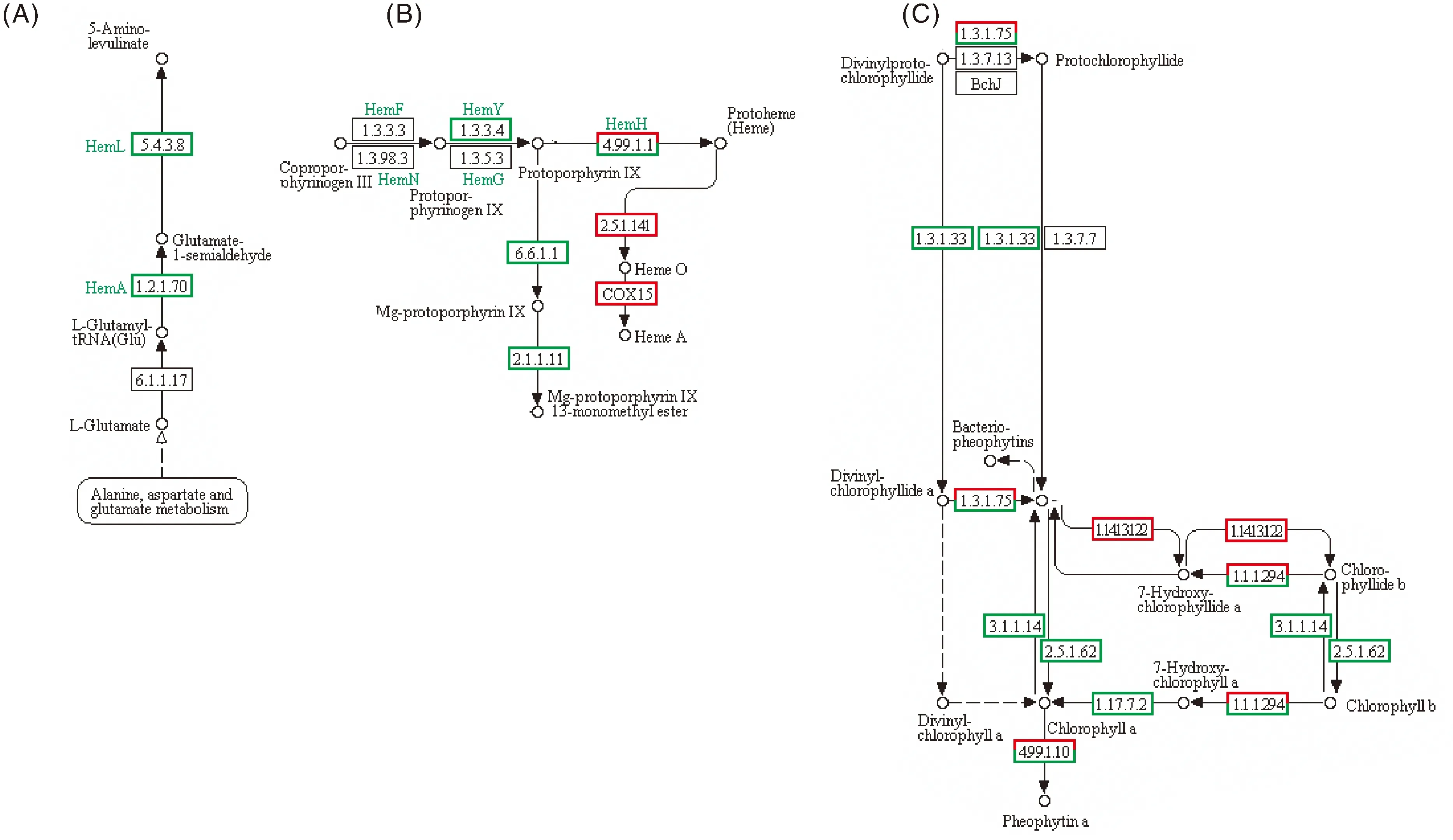
A,ALA合成;B,原卟啉Ⅸ和血红素合成;C,叶绿素合成。红色指示上调基因,绿色指示下调基因,下同。
在类胡萝卜素合成途径中,番茄红素ε环化酶(CrtL-e)、β-胡萝卜素3-羟基酶(CrtR-b)由3个上调表达的基因编码;尽管叶黄素的代谢通路中编码CrtL-e、番茄红素β-环化酶(Crtl-b)和β环羟化酶(LUT5)的基因下调(图10-A),但总体上类胡萝卜素合成相关DEGs上调表达。尤其是CrtR-b不仅在叶黄素合成中调控α-胡萝卜素和叶黄素合成,而且还广泛调控β-胡萝卜素转化成其他色素(图10-B)。CrtR-b作为调控β-胡萝卜素羟基化过程的关键酶,可能参与南瓜子叶黄化的分子调控。
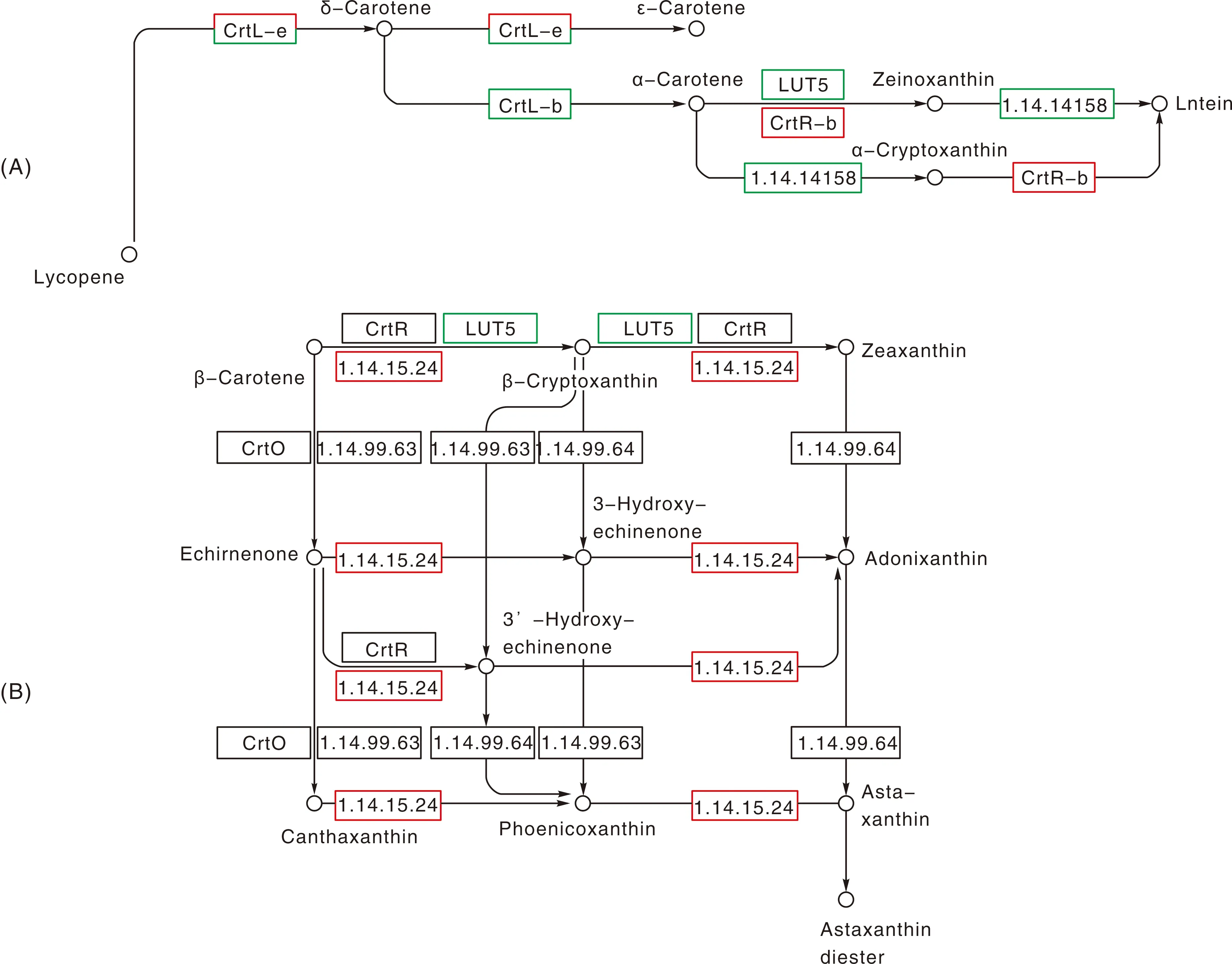
图10 类胡萝卜素生物合成途径
2.5 光合作用相关DEGs的表达
从黄化苗的光合作用途径中检测到49个关键DEGs(图11)。与光系统Ⅱ(PSⅡ)相关的27个psb基因中,11个基因呈现差异表达。psbC编码紧密结合PSⅡ反应中心的核心天线蛋白CP43,psbE编码Cytb559的α亚基,psbO、psbP、psbQ基因分别编码PSⅡ放氧增强蛋白1、放氧增强蛋白2和放氧增强蛋白3,psbR亚基对于放氧复合体的组装和稳定具有至关重要作用,psbW缺失会使PSⅡ的超分子结构不稳定,psbY基因与PSⅡ成分的其他基因如psbO、psbQ、psbS和psbTn的功能相似;psb27与放氧复合体的组装有关,psb28影响CP47合成。南瓜黄化苗子叶中这10个基因表达均下调。

图11 光合作用途径
编码光系统Ⅰ(PSⅠ)P700叶绿素a脱辅基蛋白(psa)基因家族的16个基因中,psaD、psaE、psaF、psaG、psaH、psaK、psaL、psaN、psaO共9个基因下调,这些亚基蛋白主要参与PSⅠ反应中心的组成。编码细胞色素b6f(Cyt b6f)复合物的Pet基因家族中,编码细胞色素f的PetA基因既有上调又有下调,编码Rieske Fe/S蛋白的PetC基因下调。编码电子传递过程的Pet基因家族中,编码质体蓝素的PetE、铁氧还原蛋白的PetF、铁氧还蛋白-NADP+还原酶的PetH基因既有上调也有下调表达。编码F型ATP酶的基因家族中,有2个编码F型H+/Na+转运ATP酶亚基和5个编码F型H+-ATP酶亚基的基因表达下调。
捕光色素蛋白复合体(LHC)主要参与光能吸收与传递,保证光合作用顺利进行[31]。除了LHCⅠ叶绿素a/b结合蛋白5(Lhca5)外,其余编码Lhca1-4和LHCⅡ叶绿素a/b结合蛋白(Lhcb1-7)的基因表达量均显著下降。
3 讨论
非模式植物的可用遗传资源和功能基因组学信息有限。通过高通量测序技术进行转录组分析是研究基因功能、基因表达动态、生物代谢调控机制等快速有效的方法。Zhang等[32]通过转录组分析,分别在哈密瓜冷胁迫12 d和24 d鉴定了6 479和7 914个DEGs,并通过GO和KEGG富集分析得出这些DEGs主要与淀粉和蔗糖代谢、戊糖磷酸途径和糖酵解/糖异生等相关。Zhang等[33]通过转录组分析甜瓜胚性愈伤组织和非胚性愈伤组织的基因序列,认为与光合作用、代谢途径和次生代谢产物生物合成相关的基因可能促进非胚性愈伤组织向胚性愈伤组织分化。本试验对蜜本南瓜自然黄化突变体幼苗子叶进行转录组分析,从南瓜黄化苗和对照植株中分别鉴定了25 043和25 132个表达基因,其中1 313和1 402个基因分别在黄化苗和对照中特异表达;检测到12 687个DEGs,其中上调表达的基因6 444个,下调基因6 243个。
植物光合色素主要包括Chl和类胡萝卜素。植株叶色主要取决于Chl和类胡萝卜素的相对含量,叶绿体缺失直接导致白化突变体产生,而Chl合成受阻导致Chl缺乏和类胡萝卜素含量过高的植株大多呈现叶片黄化表型[34]。叶色变化是由复杂的生物过程决定的,其分子机制可能涉及叶绿体蛋白合成突变、Chl合成受阻,以及核-质基因互作或不亲和性等[35]。参与Chl a和Chl b合成的关键酶有谷氨酰-tRNA还原酶(催化ALA合成)、镁离子螯合酶(催化Mg-原卟啉Ⅸ合成)和叶绿素a加氧酶(催化Chl b合成)。此外,由原卟啉Ⅸ第二条分支合成的血红素一方面可以通过负反馈调节Glu-TR活性进而影响ALA合成[36],另一方面其最终会生成光敏色素进而参与植物光形态建成。因此血红素代谢途径中基因突变也会造成Chl含量改变进而导致叶色变化。本试验结果表明,黄化苗在Chl合成过程中,大多数关键调控基因(如谷氨酰-tRNA还原酶、粪卟啉原Ⅲ氧化酶、镁离子螯合酶基因)呈现下调表达,因此,可能是Chl合成途径中多个反应受阻导致植株黄化突变。调节血红素合成的原卟啉铁螯合酶基因、调节血红素O合成的血红素O合酶基因、调节血红素A合成的细胞色素c氧化酶组装蛋白15亚基的基因表达均上调,表明ALA合成可能受到血红素的负反馈调节从而受到抑制。由Chl生物合成前端部分基因突变导致的叶色变化,通常呈现整株颜色变化,而由生物合成后期基因突变引发的叶色变化,通常使植株叶色呈现条纹、斑点等变化[37-38]。本试验南瓜黄化苗整株呈黄色,其可能由于hemA、hemL的表达下调引起,这与前人的研究结果一致。
叶绿体的形成是一个复杂的过程,多数叶绿体蛋白由核基因编码,再通过细胞质的加工处理,最终在叶绿体内发挥功能[39]。而这些基因突变会导致叶绿体形成相关蛋白发生改变,从而使叶绿体发育缺陷,进一步造成叶色黄化。Chl a、Chl b主要分布在叶绿体的类囊体膜上,参与光合作用的光能吸收、传递与转换。在类囊体薄膜上可分离出4类蛋白复合体:PSⅠ、LHCⅠ,PSⅡ、LHCⅡ,以及Cyt b6f复合体,F型H+-ATP酶复合体,它们嵌入高度折叠的类、囊体膜中,负责光能吸收和传递[40-41]。在光合作用途径中,PSⅠ和PSⅡ参与水裂解放氧反应和原初电荷分离等关键步骤,是生物光能转换和影响光合效率的重要场所[42]。本试验发现,在黄化叶片中psb和psa基因家族均有许多基因下调表达,推测PSⅠ和PSⅡ中相关蛋白的功能可能受阻,黄化苗光合性能下降,这与王力源等[2]的结果相似。此外,PSⅠ主要分布在基粒片层的重叠区域[43],PSⅡ蛋白复合物的显著下调可能导致不良基粒的堆叠[44]。在光合途径中,Rieske Fe/S蛋白作为组成Cyt b6f复合体的一个大亚基,由细胞核基因PetC编码,PetC缺失会导致Cyt b6f亚基严重下调,并且线性电子传递严重受阻,导致光合作用过程受到抑制。捕光复合物叶绿素a/b结合蛋白是由核基因编码的一组类囊体蛋白,通过与色素结合参与光能的吸收与传递,而植物有超过60%的Chl与该复合体结合,对光合作用有重要影响[45]。本试验光合作用-天线蛋白是KEGG富集因子最大的通路,且绝大部分LHC家族基因下调表达。因此南瓜叶片光能吸收与传递受到严重抑制。
4 结论
本试验通过转录组分析,共检测到12 687个DEGs,其中有6 444个上调,6 243个下调,通过qRT-PCR验证了转录组数据的可靠性。通过GO和KEGG富集分析发现黄化苗在Chl、类胡萝卜素合成、光合作用-天线蛋白、光合作用途径中出现大量DEGs。在Chl合成过程中谷氨酰-tRNA还原酶、镁离子螯合酶、叶绿素a加氧酶异常表达,以及血红素代谢相关基因上调抑制Chl合成可能是叶色黄化的主要原因。同时,在类胡萝卜素合成途径中β-胡萝卜素3-羟基酶促进类胡萝卜素生成。另外光合作用过程受阻也反馈抑制Chl生物合成。本试验结果为南瓜黄叶形成的分子机制提供了新的见解,为标记开发、基因克隆和南瓜分子育种奠定了理论基础。
