基于Iso-Seq技术的野生马齿苋叶片转录组分析
2023-02-21叶梅荣黄守程王晓鹏刘爱荣
叶梅荣,黄守程,王晓鹏,刘爱荣,崔 峰,康 健
(安徽科技学院 生命与健康科学学院, 安徽 凤阳 233100)
全长转录组测序(isoform sequencing,Iso-Seq)是指利用三代测序技术获得生物体内完整的RNA分子的全长序列,建库过程中RNA分子无需打断,测序可直接获得完整的cDNA序列[1]。第三代测序技术可以获得更长乃至全长的转录组[2]。三代单分子测序技术的应用降低了无参考转录组的分析难度,能够较为容易地获取完整基因组和全长转录本,对深入研究生物体的转录机制极有利。三代测序是无需PCR扩增、操作更加简单、通量高、速度快、读长碱基对的新型测序技术,但比二代测序错误率高,因此结合三代和二代测序技术更有利于研究生物体的转录组[3]。三代测序技术刚兴起,但是发展很快,如Kuang等[4]利用Iso-seq探讨东北红豆杉(TaxuscuspidateSieb. et Zucc.)的转录组,揭示了东北红豆杉生物合成的复杂性。
马齿苋(PortulacaoleraceaL.)别称有马苋、五行草、长命菜、五方草、马齿菜等,为马齿苋科 (Portulacaceae)马齿苋属(Portulaca)[5],分布地很广,全球各地均有分布,被列为世界第八大普通的植物[6]。马齿苋既可入药,又可食用,是药食两用的野生植物。近年的药理研究表明,马齿苋具有抗菌、降血脂、抗衰老、松弛肌肉、抗炎、镇痛,以及促进伤口愈合等功效[7-9]。马齿苋营养丰富,富含多种矿质元素和多种有机营养保健成分如多糖、总黄酮、α-亚麻酸、β-胡萝卜素、ω-3脂肪酸、萜类、生物碱、去甲肾上腺素、褪黑激素等,具有改善血液循环、提高人体免疫力、防治心血管疾病、抑制微生物生长等多重功效[6-8,10]。马齿苋适应性极强,对高温、干旱、高湿、高盐、重金属污染等逆境的抵抗能力强大,是优良的生态修复植物[11-14]。马齿苋集保健、食用、药用、生态价值于一身,具有进一步研究开发的价值。
马齿苋耐高温和干旱,不耐低温,因此本研究利用干旱和低温胁迫分别处理马齿苋,同时设置对照,目前马齿苋没有参考基因组,因此本研究基于三代(Iso-Seq)测序技术和二代测序技术研究马齿苋叶片的全长转录组,以期为马齿苋属的基因研究提供参考。
1 材料与方法
1.1 材料
马齿苋种子播种于花盆中(盆高14 cm,下口直径11 cm,上口直径23 cm),基质是通用型“中禾”营养土,光照培养箱培养温度白天30 ℃,晚上25 ℃。待种子萌发成幼苗后,每盆留有5株幼苗。待苗长至5 cm 高度左右,设置低温处理(LTS,白天10 ℃,晚上5 ℃)和对照处理即常温处理(C,白天30 ℃,晚上25 ℃),同时设置自然干旱处理(DS,白天30 ℃,晚上25 ℃,即一次浇透水后,连续14 d不浇水),对照组(即常温处理组)每7 d浇透1次水,每个处理重复3次。低温处理和干旱处理14 d后,分别取各处理的马齿苋成熟叶片用液氮速冻,然后干冰运送到武汉贝纳科技服务有限公司进行三代和二代转录组测序和分析。马齿苋在培养过程中因处理时间较长已进入开花结实阶段。
1.2 总RNA提取、cDNA文库构建和二代技术转录组测序
提取马齿苋样本的RNA,用琼脂糖凝胶电泳检测RNA质量,用NanoDrop ND-1000超微量分光光度计检测RNA的纯度和浓度,采用Agilent2100生物分析仪精确检测RNA的完整性,利用Illumina HiSeq 2500测序平台进行高通量测序。
1.3 文库构建和三代技术转录组测序
利用Oligo(dT)富集带有PolyA尾的RNA,使用SMARTerPCR cDNA Synthesis Kit将RNA反转录合成cDNA,使用KAPA HiFi PCR Kits进行PCR扩增合成cDNA;利用BluePippin进行片段筛选,用SMRTbell template prep kit 1.0构建SMRTbell文库,进行损伤修复,末端修复;在DNA片段两端连接茎环状测序接头,并利用外切酶去除连接失败的片段。文库定量后,利用PacBio Sequel Ⅱ对cDNA文库测序,获得原始下机数据(polymerase reads)。
1.4 数据质控
对polymerase reads进行过滤,去除adaptor序列、长度小于50 bp和准确度小于80%的polymerase reads,然后计算合格的插入片段(subreads)的数目、长度分布和N50,利用CCS软件校正获得高质量的插入片段序列(reads of insert,ROI)即一致性序列(Circular consensus sequencing,CCS),利用smrtlink 5软件对插入片段的接头序列进行识别和去除,得到初级转录本,初级转录本存在大量的冗余序列。利用ICE(isoform-level clustering algorithm)算法将冗余序列聚类到一起,得到新的一致性序列,最终得到一致性序列。再利用CD-HIT(version4.6.7)软件去除冗余序列分别得到isoform级别序列,将isoform级别序列继续去除冗余序列得到unigene。所有数据已上传美国国家生物技术信息中心(BioProject: PRJNA734447)。
1.5 Unigene功能注释
将universal gene(unigene)与universal protein(Uniprot)、Pfam protein families(Pfam)、gene ontology(GO)、kyoto encyclopedia of genes and genomes(KEGG)、evolutionary genealogy of genes: non-supervised orthologous groups(eggNOG)、KEGG pathway(pathway)和non-redundant protein(Nr)数据库进行同源比对,获得所有注释信息。并对注释到各数据库的信息功能注释和分类。
1.6 简单重复序列标记(simple sequence repeats,SSR)信息分析
对unigene进行SSR(Simple Sequence Repeats)位点查找,设置参数为:(1/10)(2/6)(3/5)(4/5)(5/4)(6/4),其中(3/5)代表3个碱基重复5次或以上,以此类推。
1.7 差异表达基因筛选
二代原始测序数据经过过滤得到clean reads,因马齿苋没有参考基因组,因此与三代测序得到的全长非嵌合的转录本经CD-HIT去冗余后得到的isoform作为参考序列进行比对,用RSEM对比对结果进行统计,以FPKM(fragments per kilobase of transcript per million fragments mapped)≥1为标准筛选有效表达基因,利用DEGseq软件,以Q value<0.05,|log2FoldChange|>2为标准筛选差异表达基因(differentially expressed genes,DEGs)。
2 结果与分析
2.1 初级转录本
为了得到尽可能多的转录本,将低温处理、干旱处理及对照组(每个处理重复3次)共9个样本的叶片混合一起,反转录成cDNA,总共得到641 732条原始片段,N50为144 732,质控后得到插入片段26 758 071,总碱基量为44 G,其N50为2 002,利用CCS软件质控得到高质量的插入片段442 760条(表1),N50为2 397,说明测序质量较好。利用smrtlink软件对高质量的插入片段进行识别和去除接头序列,得到全长的插入片段和全长的去除嵌合体的插入片段分别为391 258条和301 412条,全长片段所占比例越高,说明测序质量越好。通过ICE算法去除冗余序列得到188 792条一致序列。利用CD-HIT软件将相同转录本聚类到一起,得到103 298条isoform序列。

表1 初级转录本统计
2.2 Unigene级别聚类分布
根据CD-HIT软件进一步将来源于一个基因的isoform聚类到一起,即从103 298条去除冗余得到39 717条unigene序列(图1)。unigene集中分布在600~3 200 bp,所占百分比为88.34%。
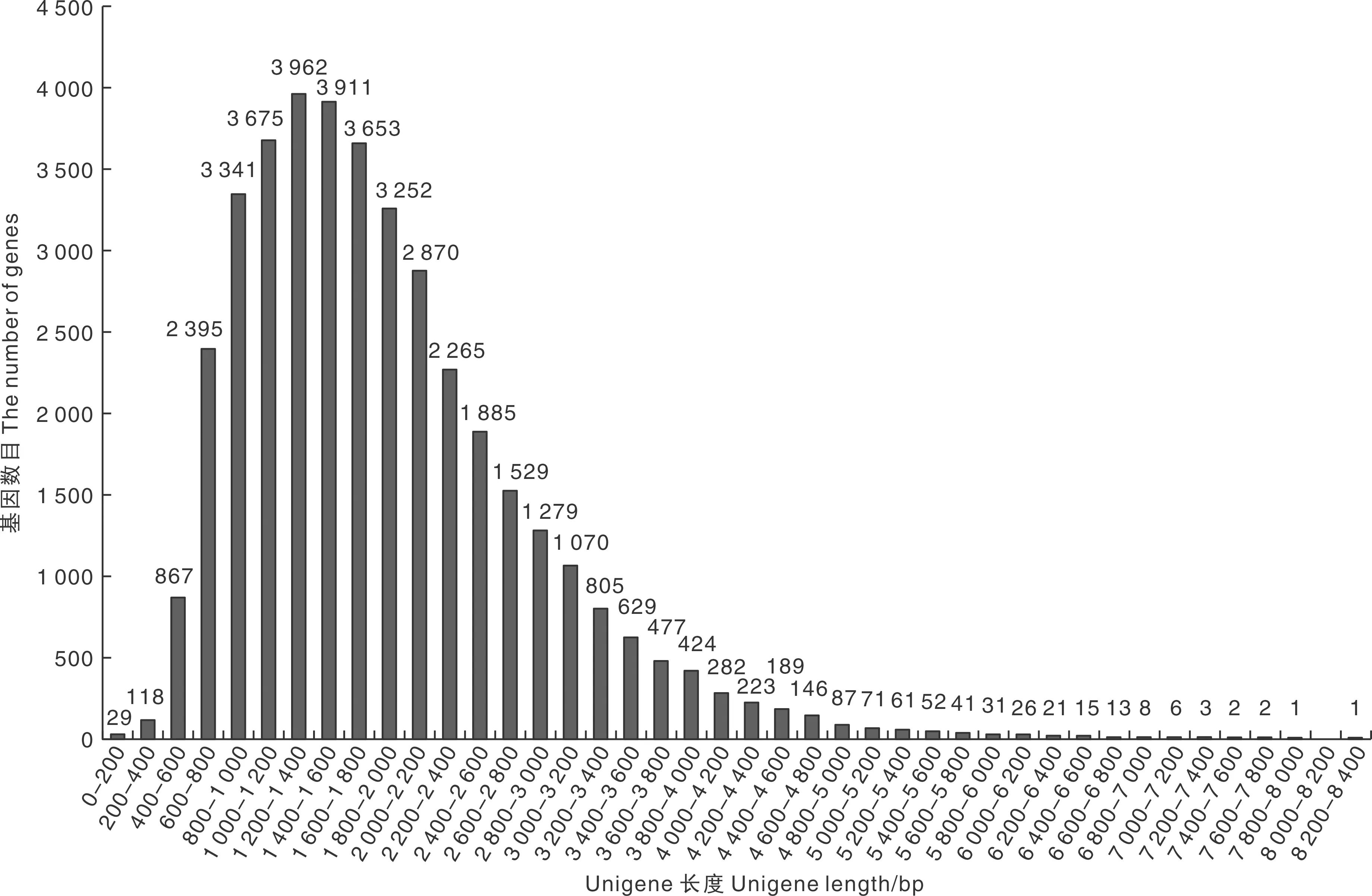
图1 Unigenes长度分布图
其中1 200~1 400 bp的unigene数量最多,具有3 962条,占百分比为9.98%;其次是1 400~1 600 bp的基因,有3 911条,占百分比为9.85%。
2.3 LncRNA预测
LncRNA(长链非编码RNA)是一类转录本长度超过200核苷酸(nt),不编码蛋白质的RNA分子。根据核心程序plek采用ncrna_pipeline流程对得到的去冗余转录本序列预测LncRNA,得到40 952条没有编码能力的转录本序列,长度主要分布在400~2 200 nt,占比95.10%。其中长度600~800 nt之间分布数量最多,为7 636条,所占百分比为18.65%;其次是长度为800~1 000 nt的基因,有7 514条,占比为18.35%(图2)。
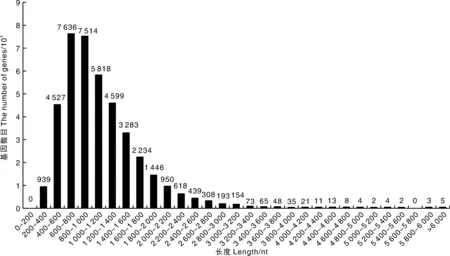
图2 LncRNA长度分布图
2.4 ORF预测
开放阅读框(open reading frame,ORF)是蛋白质编码基因的正常核苷酸序列,从起始密码子到终止密码子的阅读框可编码完整的多肽链,期间不存在使翻译中断的终止密码子。利用transDecoder(版本:5.5.0)对unigene进行读码框预测,鉴定标准为ORF长度大于300 bp,该序列似然函数的log值大于0,选取6个ORF中分数最大的一个,选择最长的一个,一共预测出38 419条CDS序列,其长度分布图见图3。从图3可以看出,CDS集中分布在200~1 600 bp,占比为83.25%,其中分布最多的是200~400 bp的条带,有7 464条,占比为19.43%;其次为400~600 bp的条带,有6 342条,占比为16.51%。
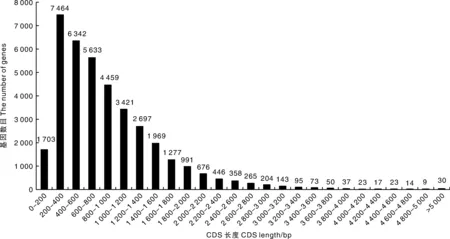
图3 CDS长度分布
2.5 Unigene的功能注释
将样本unigene与Nr、Uniprot、Pfam、GO、KEGG、eggNOG和Pathway数据库进行同源比对,被注释的基因有36 381条,注释率为91.6%;未注释的基因有3 336条。注释率最高的为Nr,注释了36 034条基因,注释率为90.73%;其次是Uniprot,注释了29 464条基因,注释率为74.18%(图4)。
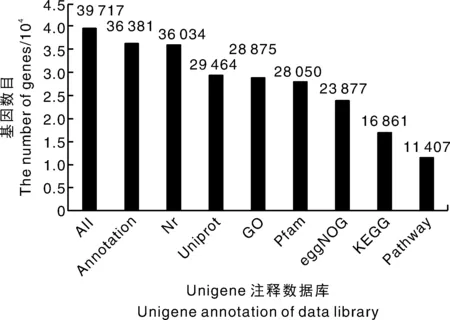
图4 Unigene 条目的注释统计
2.5.1 GO分类
对注释上GO分类的28 875个基因进行功能的预测(图5),主要分为3大类:分子功能、细胞组分和生物学过程,这3大类最多的分别是离子结合、细胞组分和生物学合成过程,分别有11 385、8 272和8 055个基因。
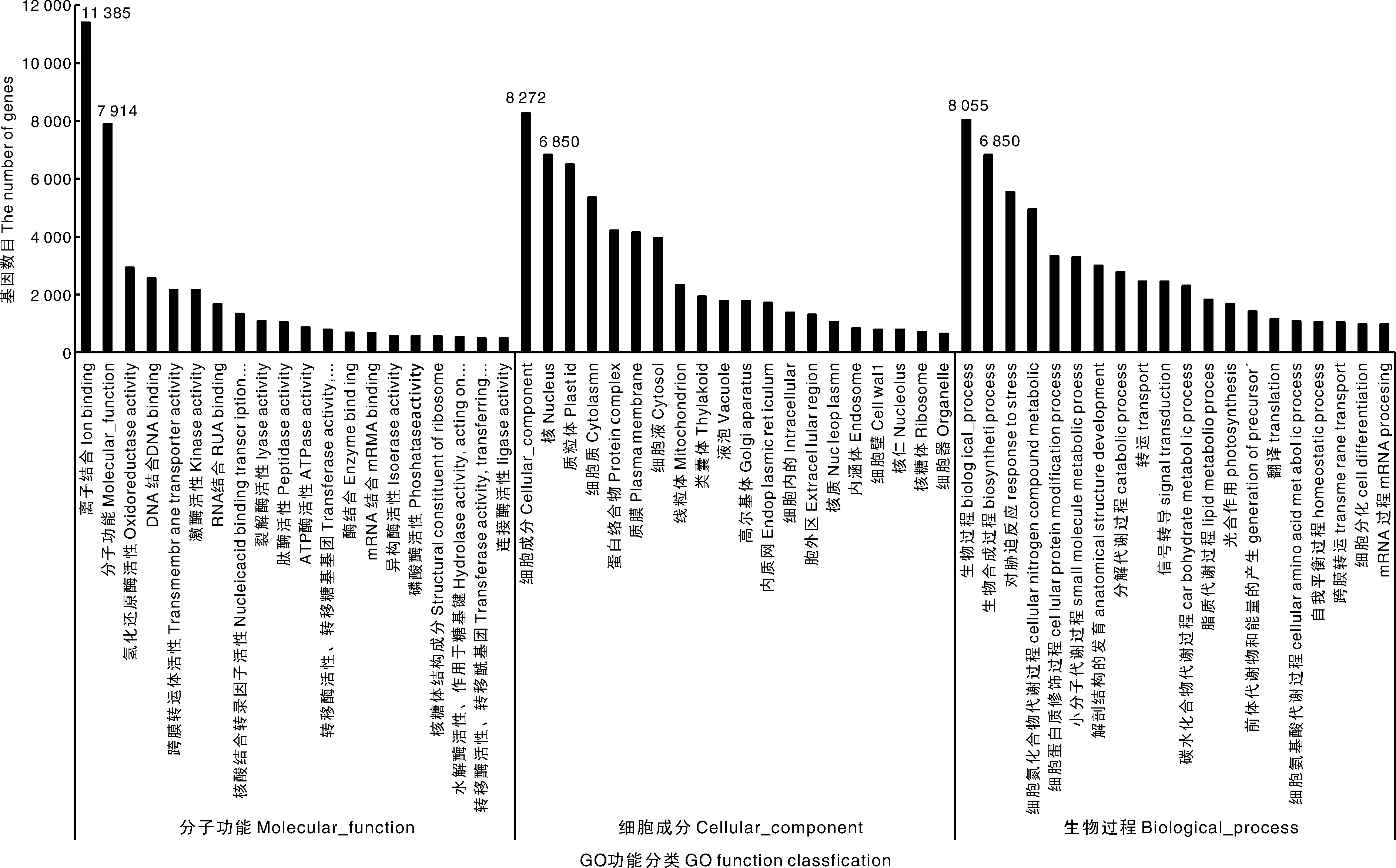
图5 GO功能注释
2.5.2 KEGG pathway分类
对注释上KEGG的16 861个基因进行代谢通路分类(图6),总共分为5大类,包括有机系统、代谢、遗传信息过程、环境信息过程和细胞过程;这5大类程基因数量最多的分别为内分泌系统、碳水化合物代谢过程、翻译、信号转导及运输和分解代谢,其基因数量分别为1 057、3 105、1 521、2 442和1 109个。从图中还可以看出代谢所占基因数最多,其占总注释基因数55.25%。
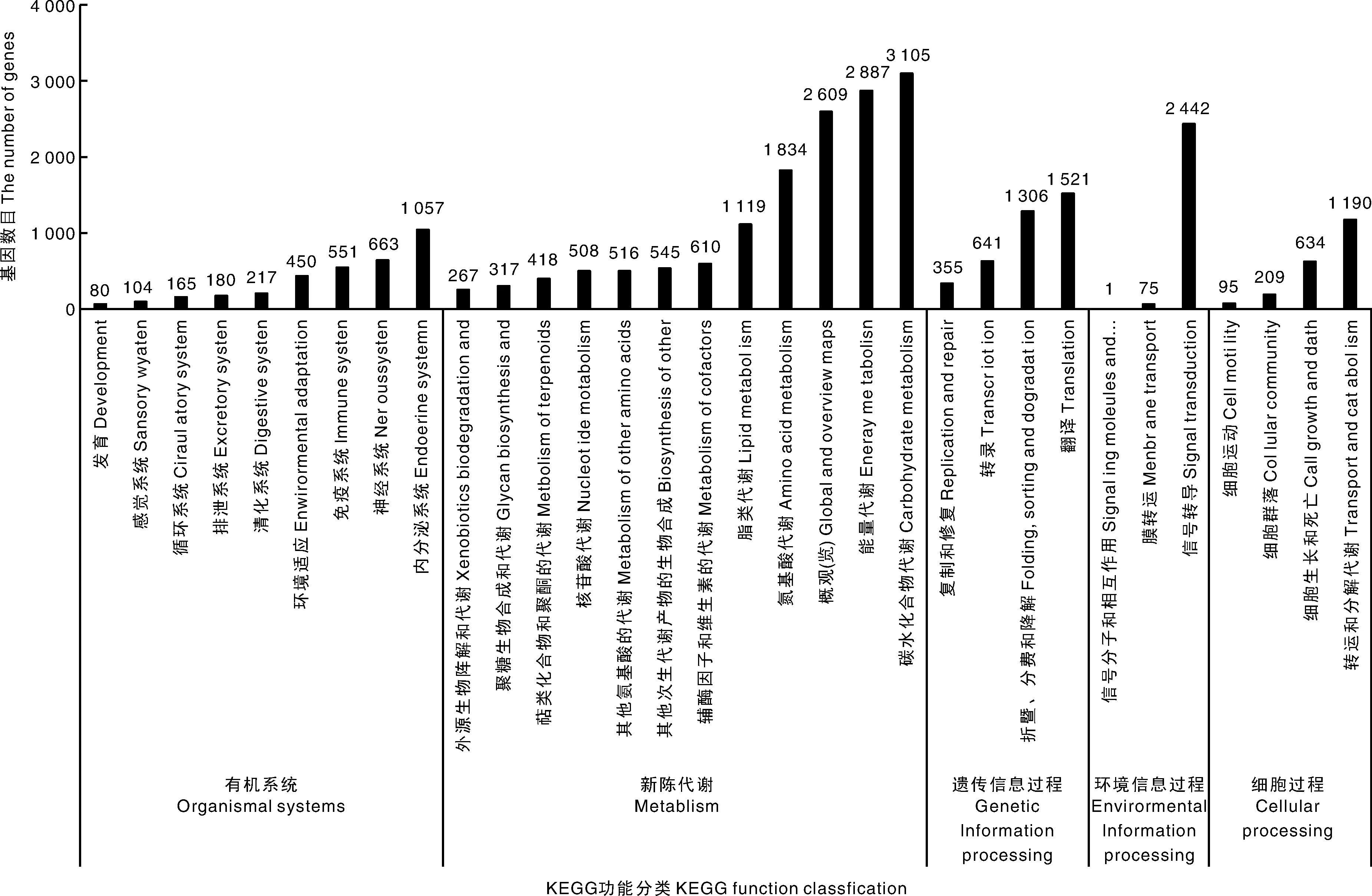
图6 KEGG功能注释
2.5.3 COG功能分类
利用COG对基因进行直系同源分类,分为26个组(图7),然后对unigene进行功能统计,结果发现信号转导机制所占基因数量最多,为1 318,其次是翻译、核糖体结构和生物发生,为1 236个基因,排第三的是翻译后修饰、蛋白折叠和蛋白伴侣,为1 234个基因。
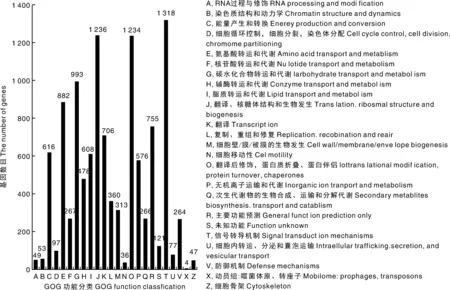
图7 COG功能注释分类
2.5.4 Nr注释分类
将Unigene序列比对到Nr的蛋白序列,进而得到每个基因的Nr注释,通过Nr库比对注释的结果,统计并绘制比对上的物种分布图(图8),只统计比对最多的前10个物种,其余的划到Other species。从图8中得出比对最多的前3个物种分别是栽培甜菜、藜麦和菠菜,说明马齿苋和这3个物种蛋白序列相似度高,而划到其他物种的有9 355个基因,暗示马齿苋有自己特有的蛋白序列。且注释的E-value值在0~1×10-150、1×10-150~1×10-100和1×10-100~1×10-50占比分别为25.65%、18.56%和28.77%,说明可信值较高(图9);一致性鉴定发现一致性非常高,一致性鉴定大于60%的基因数量为27 490个(图10)。
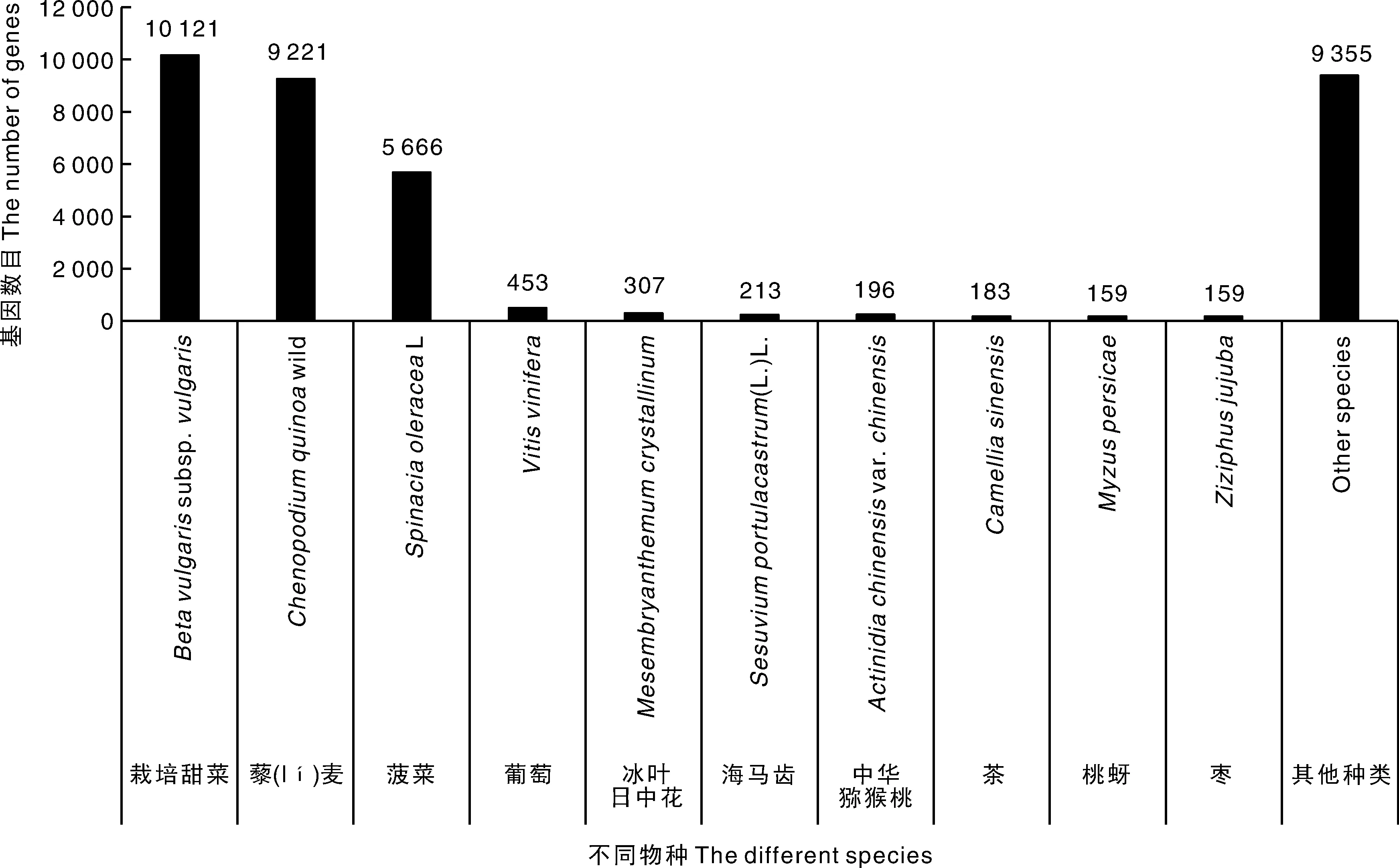
图8 Nr注释
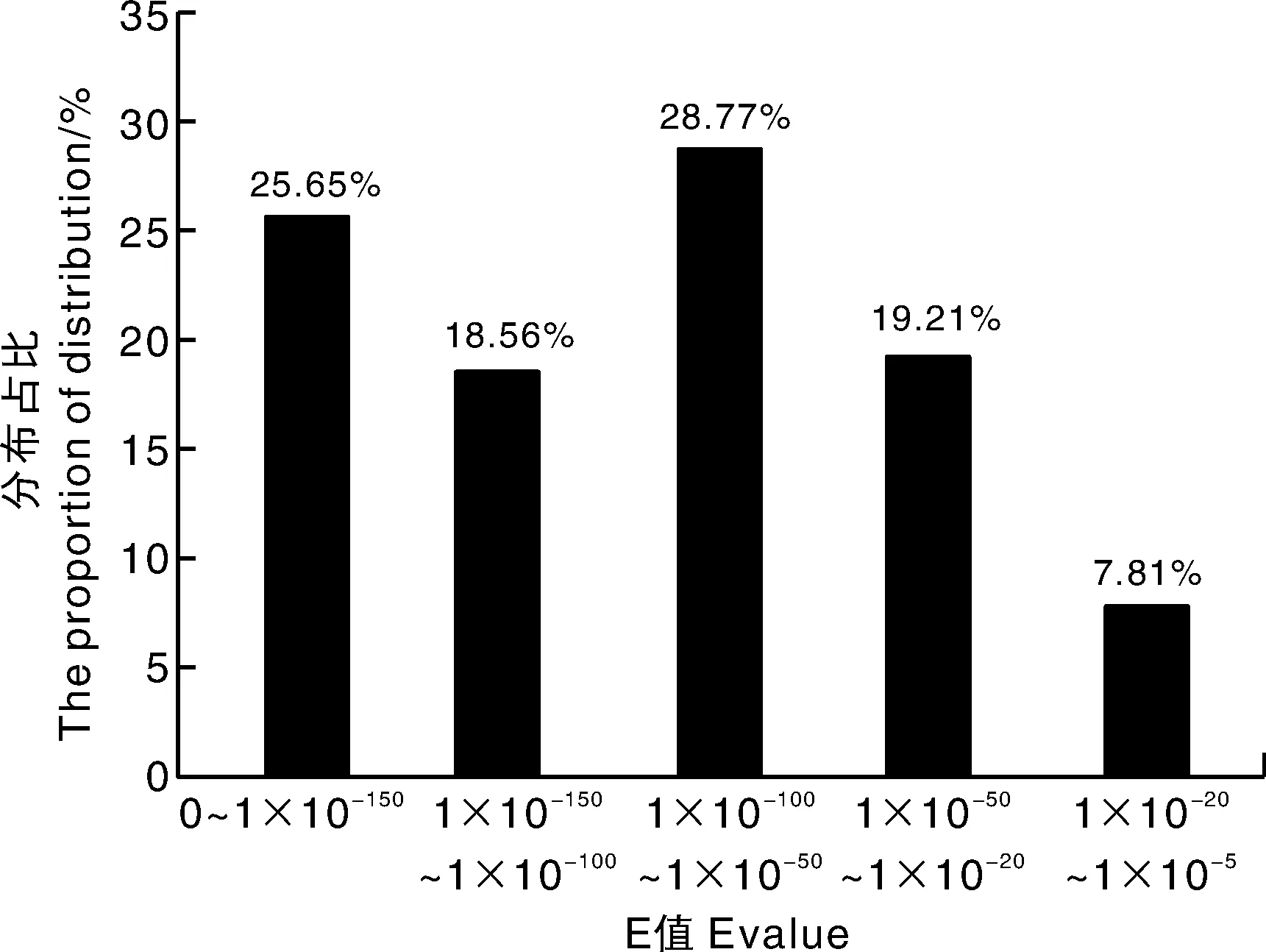
图9 Nr注释的值
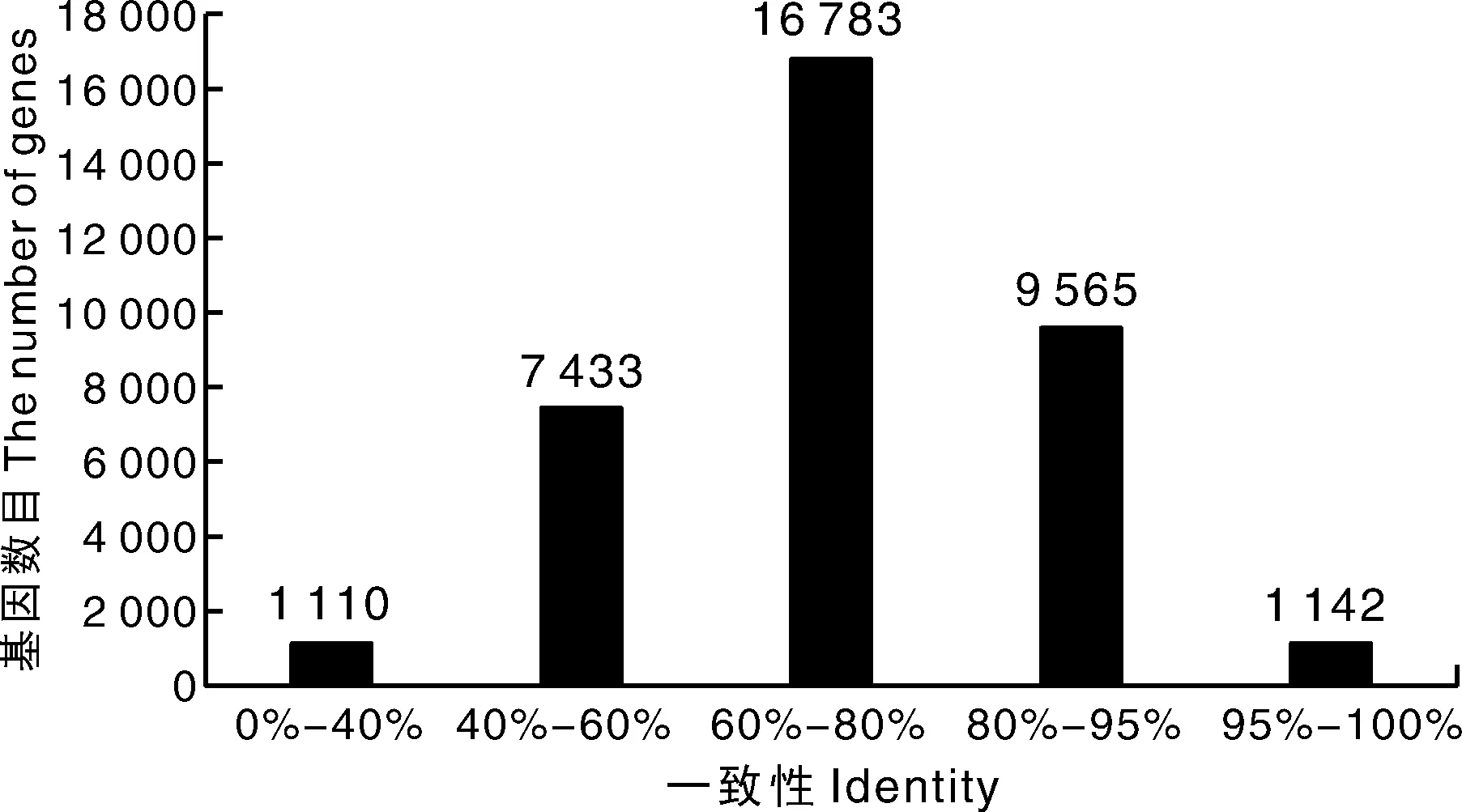
图10 Nr注释的一致性分布
2.5.5 Pfam结构域分类注释
Pfam数据库是一个蛋白质家族大集合,根据多序列比对和隐马尔可夫模型,利用hmmscan进行结构域预测,图11显示注释最多的前20个域,其中蛋白激酶(pkinase)最多,有1 080个基因,其次是酪氨酸蛋白激酶(Pkinase_Tyr),有1 027个基因。
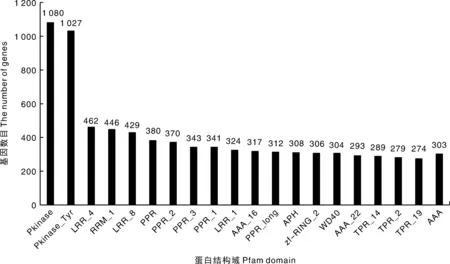
图11 Pfam结构域分类注释图
2.5.6 SSR鉴定
图12显示unigene总共鉴定了25 898个SSR位点,其中单个碱基重复的最多,为12 772个,2个碱基重复的位列第二,为5 961个,排在第三的为3个碱基重复序列,是5 842个。
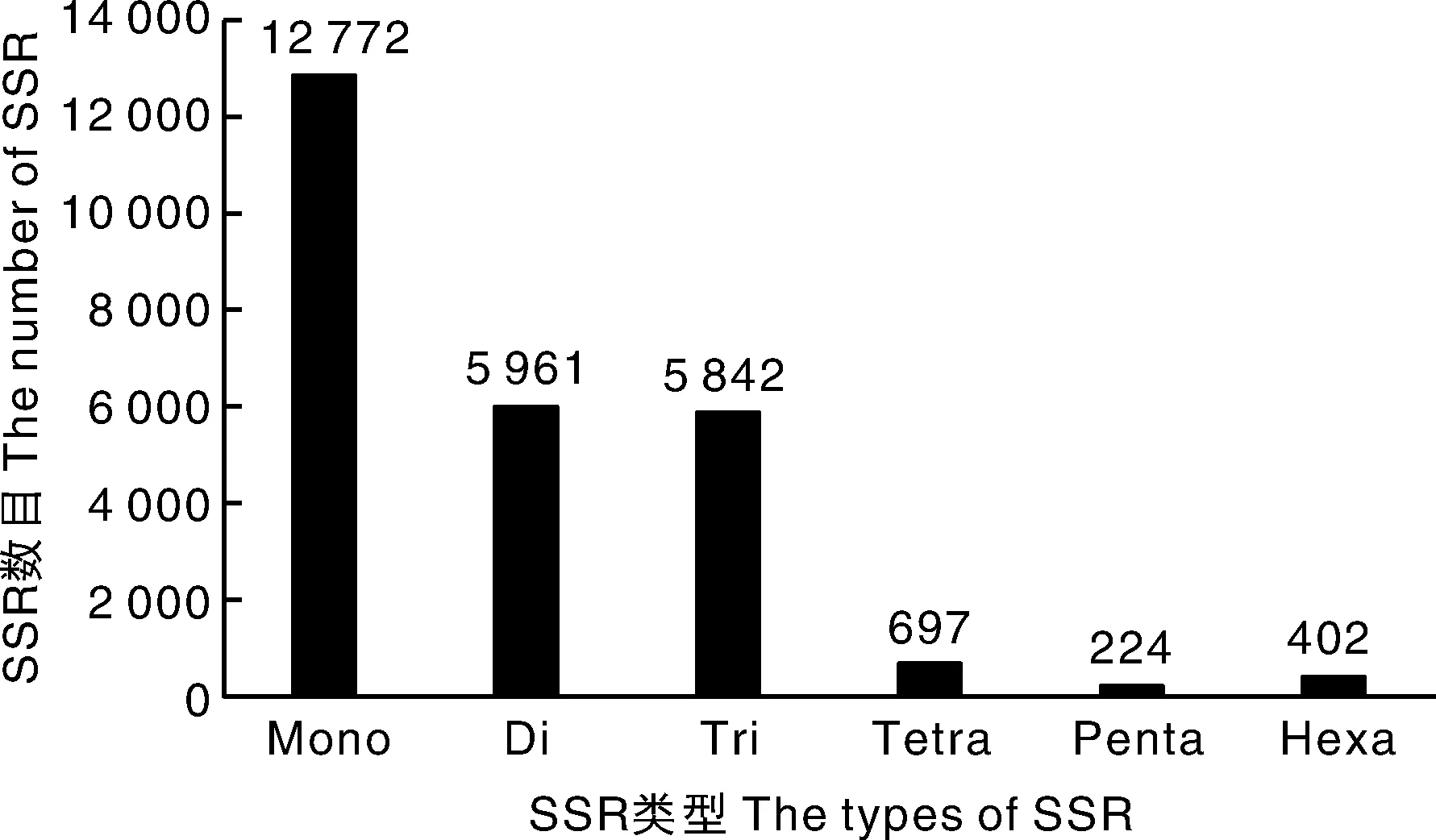
图12 SSR重复单元数
2.6 差异表达转录本的筛选
根据差异表达分析软件DESeq2筛选差异表达基因(DEGs),筛选标准为:如果Qvalue<0.05,且log2FoldChange>2,则DEGs被认为是显著上调差异表达基因;如果Qvalue<0.05,且log2FoldChange<-2,则DEGs被认为是显著下调差异表达基因。差异基因火山图可以显示不同处理前后的差异基因分布,其中用红色表示显著上调的基因,以绿色表示显著下调的基因,以黑色表示无显著性差异的基因(图13-A、13-B、13-C)。结果显示,C vs DS之间鉴定出16 194个DEGs,其中包括2 874个上调DEGs,13 320个下调DEGs;C vs LTS之间获得了16 093个差异表达基因,其中包括6 365个上调DEGs,9 728个下调基因;DS vs LTS之间发现了25 498个DEGs,其中包括15 163个上调DEGs,10 335个下调DEGs。通过维恩图比较不同处理比较组的3个数据集(C vs DS、C vs LTS、DS vs LTS),在3个比较组中,共有2 430个DEGs(图13-D),除3个比较组共有的DEGs外,在C vs DS和C vs LTS中还共有2 656个DEGs,C vs LTS和DS vs LTS中还共有7 919个DEGs,C vs DS和DS vs LTS比较组中共有8 195个DEGs,且每个比较组中都有自己特有的差异表达基因,C vs DS、C vs LTS、DS vs LTS中特有的差异表达基因分别有2 913、2 088和6 954个(图13-D)。
2.7 差异表达基因GO功能分析
图14分别列出前20个富集最显著的差异表达基因,C vs DS差异基因数量最多的是赤霉素介导的信号转导途径,有77个;其次是一个氧原子结合的氧化还原酶活性,有66个;排在第三的是钙离子介导的信号转导途径,有54个(图14-A);C vs LTS差异基因主要表现在细胞对各种光的反应、叶绿素合成过程与丙酮酸磷酸二激酶活性等方面(图14-B);DS vs LTS差异基因数目排列前三的分别为丙酮酸磷酸二激酶活性(222个)、木质素生物合成过程(有127个)、叶绿体核质体(118个)(图14-C)。
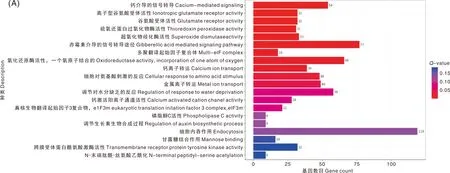
图14 C vs DS (A), C vs TS (B), DS vs LTS (C)差异表达基因GO分类图
3 结论与讨论
马齿苋被世界卫生组织列为最常用的药用植物之一,是世界上第八大普通植物,被我国卫生健康委员会划定为78种药食同源的野生植物之一,生态适应能力强,抗旱抗涝、耐盐碱,在我国野生资源丰富,但未被充分重视和利用。因此,本文研究常温(对照)、干旱胁迫和低温胁迫下的野生马齿苋叶片的全长转录本,并进行功能注释和GO分类等研究,以期为加强马齿苋的利用和其基因组的研究提供参考。根据测序共得到39 717条unigene,通过与Nr、Uniprot、Pfam、GO、KEGG、eggNOG和Pathway数据库进行同源比对发现,被注释的基因为36 381条,未被注释的有3 336条,暗示马齿苋有未知的转录本,可为马齿苋属植物新转录本的研究提供参考。本研究发现单个SSR位点数最多,为12 772个,2个位点和3个位点的SSR分别为5 961和5 842个。SSR标记具有多态性高、信息量大等特点,常被用于物种的遗传多样研究、遗传图谱的绘制等,因此本转录组的SSR数据可以为后续马齿苋属植物的研究提供参考。差异表达基因筛选结果表明,干旱和低温胁迫处理中上调和下调的差异表达基因数量较多,且不同比较组中除了有共有差异表达基因外,还有自己特有的差异表达基因,这些为以后研究这些基因的功能提供了参考依据。
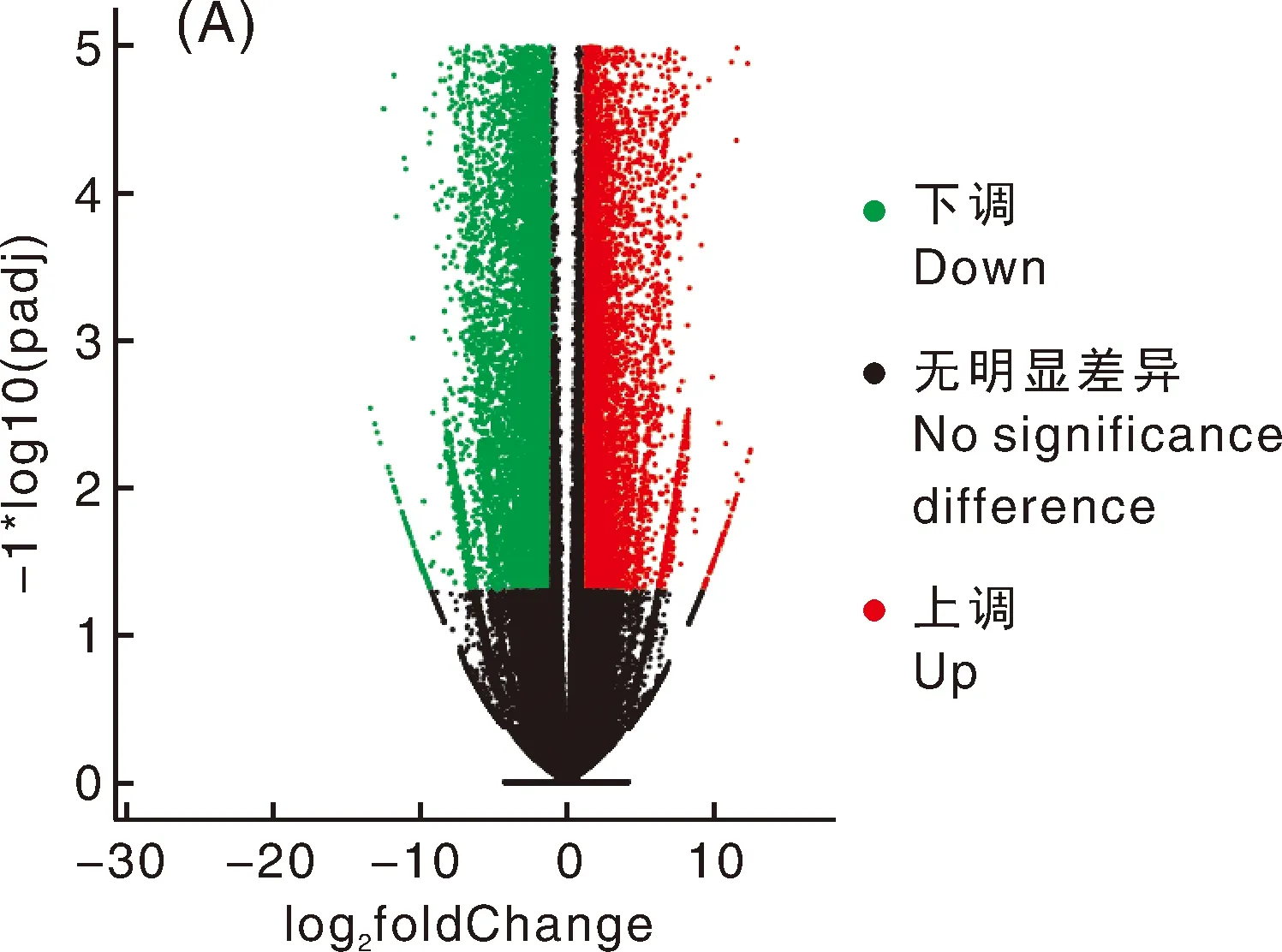
A,C vs DS 火山图;B,C vs LTS 火山图;C,DSvsLTS 火山图;D,不同处理组比较的维恩图。
A, C vs DS Volcano; B, C vs LTS Volcano; C, 15 DSvsLTS Volcano; D, Venn diagram of DEGs in different comparisons.
图13差异表达基因分布
Fig.13Distribution of differentially expressed genes
