CASTEP在电子结构教学中的应用
2023-02-21宋宏权陈培佳
宋宏权 陈培佳 刘 婧 张 薇
周口师范学院物理与电信工程学院 河南周口 466001
固体物理这门课程的主要研究对象是晶体,研究了固体的结构及其组成粒子间的相互作用与运动规律,该研究的主要目的是为以后新型材料的研究提供帮助[1]。不同固体材料的物理化学性质都存在着不同,这是因为它们的电子结构及电子的运动存在着区别,由于电子结构在固体物理学中所涉及的内容广泛且较为抽象,这不仅要求学生有一定的物理思想,还要有扎实的数学功底[2]。这都使得学生感觉学习起来比较困难,也就给电子结构内容在固体物理教学中带来较多的困难,但当学生能够通过某种手段构建出清晰的三维物理图像时,对固体物理材料的结构就会有一个全新的认识,学习电子结构将变得简单轻松了,这样做能取得事半功倍的效果[3]。随着互联网和计算机技术的发展,完全依靠课本也越来越不适合现代教育的发展,教学也要紧跟时代的步伐,适当进行改革,让学生在轻松愉快的氛围中学习知识[4]。
Materials Studio软件包括了多个计算模块,其中CASTEP模块主要用于计算能带结构、DOS、光学性质等,这个软件由国外的一个实验室小组研究发明[5]。Materials Studio[6]是常用的分子模拟软件,它已经被广泛应用到科研工作中,并且得出了很多重要的研究成果,同样,我们也可以将这种软件灵活地运用到教学工作中。通过Materials Studio软件可以显示出一些材料的三维空间结构,使得图形立体化和结构动态化,直观形象地让学生看到晶体的内部结构,通过电子行为的变化来研究物体的宏观特性[7],深刻体会其物理内涵,这样降低了学习电子结构内容的难度。
目前,已经有很多学者对于一些金属的电子结构与光学性质进行了研究。2011年王天兴等人[8]研究了掺杂前后ZnO晶体的光学性质,解释了掺杂对于体系电导率和介电函数的影响;2018年李丹丹等人[9]对不同浓度的Pr掺杂纤梓矿ZnO体系进行了研究;薛晓峰[10]研究了本征ZnO体系,Mg、Cd单掺杂与共掺杂ZnO体系,分析了各体系的电子结构和光学性质,研究发现Cd单掺杂ZnO会使晶胞体积变大。本文将结合实例介绍如何利用CASTEP模块进行固体物理学课程中电子结构相关内容的计算,对一些元素的晶体结构进行优化,分析其光学性质,希望得出的结论能为后续类似研究提供帮助。
1 计算方法和模型
1.1 计算方法
为了便于计算,我们可以把第一性原理中的密度泛函理论与Materials Studio软件有效结合。HSE06是目前计算电子结构最为准确地交换关联函数,因此本文在计算的时候选用HSE06杂化泛函。用CASTEP模块对优化后晶体的电子结构和光学性质进行计算与分析。
电子结构计算的基本步骤:先在“CASTEP”模块的“Calculation”中选择“Setup”页面,在“Task”栏中选择“Properties”,选用HSE06密度泛函,然后选择需要计算的具体内容,最后在“Analysis”中进行分析。操作步骤如图1。

图1 能带和态密度的计算
如果要对晶体的光学性质进行计算,只需在电子结构计算中的“Properties”页面下选择“Optical properties”,最后在“Analysis”下选择计算的内容即可得到结果。操作步骤如图2。

图2 光学性质的计算方法
1.2 建立模型
目前建立晶体结构的方式有很多种,文中利用Materials Studio软件中的建模模块,在MS的主界面创建一个新的project,点击file,在structures中找到需要的晶体结构并导入,打开后即可得到晶体结构。下面列出的分别是fcc、bcc、金刚石、SiC、AlNi晶体结构图。

图3 为fcc晶体,bcc晶体,金刚石晶体,SiC,AlNi结构图
2 能带结构
利用CASTEP模块计算得出一些固体材料的能带图,通过分析能带图可以得到很多有效信息:通常为了计算结果的准确性,我们会选用HSE06杂化泛函。能带图的横坐标是选取的布里渊区高对称点,可以根据导带底与价带顶的位置来判断本征半导体的跃迁方向,如果不是在同一高对称点上就是间接跃迁,例如Si就是间接跃迁。最后,可以对能带图的整体进行分析,不同的能带图的宽窄是不同的,能带越宽表明处于这个带中的电子有效质量越小[11]。
从图4的能带图中对比分析可以观察到,Cu、Al、Mg的能带分散在整个布里渊区域,而且导带有一部分穿过费米能级,验证了Cu、Al、Mg的金属性,而Al与Cu具有相同的晶体结构,因此在能带结构图中选取的高对称点是相同的。AlNi、Ti的能带结构图形类似,在费米面处能带分布密集,两者都是金属体系,相对导带而言,价带分布略微稀疏,在远离费米面处为平滑的直线,Mg和Ti的晶系对称性一样,所以选择了相同的布里渊区高对称点,图上所涉及的所有金属都没有禁带,导带与价带是重叠的,其中的电子都是载流子,有良好的导电性。

图4 为采用HSE06方法计算的Al、Cu、Mg、Ti、AlNi的能带图
通过图5分析Si、SiC、Ge的能带图,可以得出三者均为半导体。能带图的横坐标是布里渊区高对称点,这是根据本征半导体的结构所选取的,根据能带图的横坐标我们可以判断出本征半导体的跃迁方向,判断方法就是观察导带底与价带顶对应的横坐标,其中SiC的跃迁方向为X—G,跃迁类型为间接跃迁;Si的跃迁方向为与SiC的完全相同;Ge的导带最低点与价带的最高点对应的横坐标相同,都在点G处,跃迁类型与SiC、Si的不同,属于直接跃迁,所以Ge是直接带隙半导体;Si、SiC属于间接带隙半导体,与Si、SiC、Ge的能带图对比,从C的能带图中可以发现,C的带隙值为5.377eV比较大,是绝缘体。

图5 为采用HSE06方法计算的Ge、Si、SiC、C的能带图
3 复介电函数
介电函数是媒质在外加电场中对外加电场的响应,固体材料的能带结构及光学性质可以通过介电函数反映出来。介电函数的公式为:ε(ω)=ε1(ω)+iε2(ω),实虚部所决定的物理量是不同的,虚部主要影响晶体的光谱吸收能力,决定其吸收光的这一特性,实部则反映了可以储存磁场的能力,根据所反映的信息,下面来分析一些晶体的介电函数,分别如图6所示:
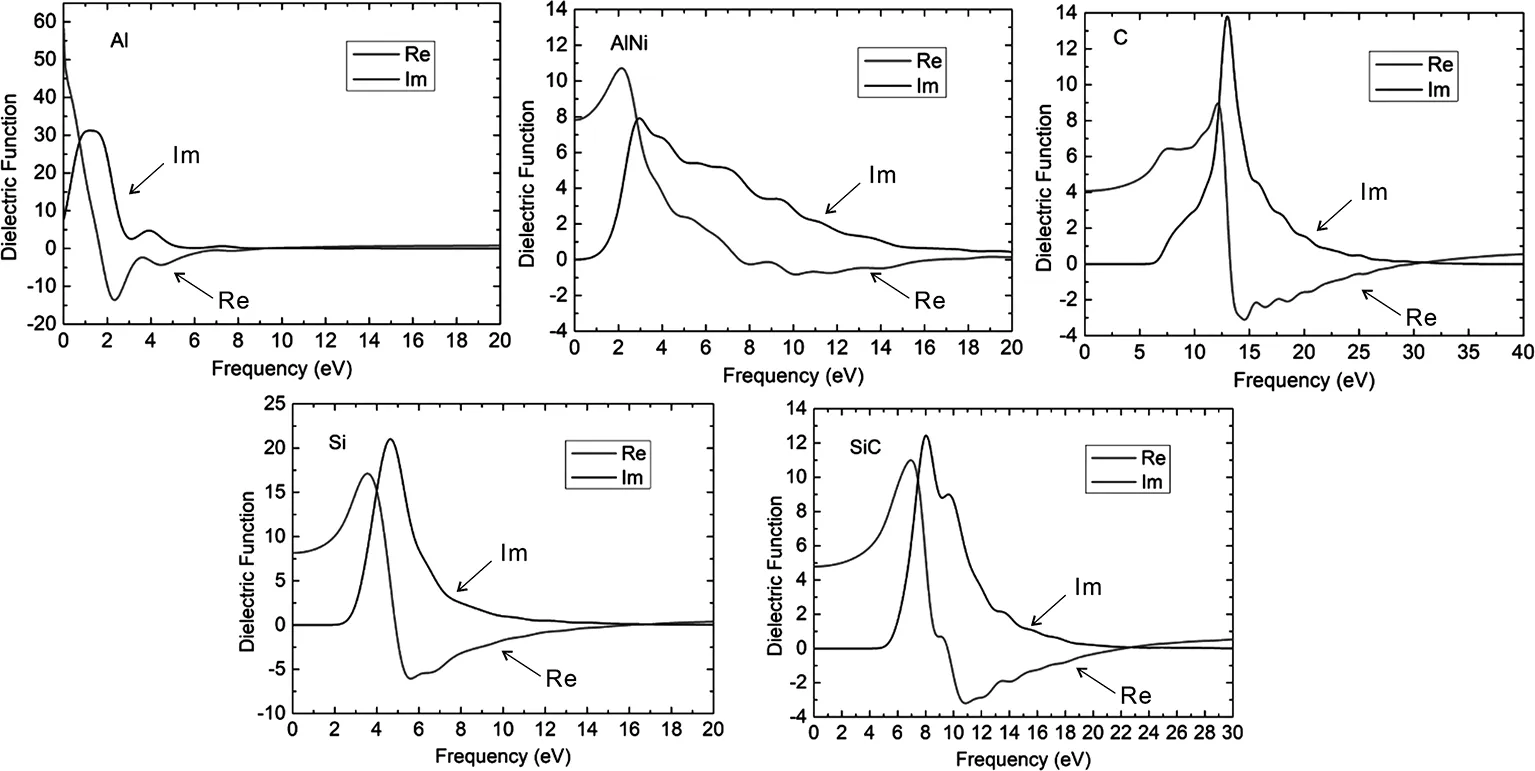
图6 为Al、AlNi、C、Si、SiC的复介电函数图
图6的横坐标代表的是入射光能量,Re曲线与Im曲线代表的是复介电函数实虚部。当入射光线的能量变为0时,其Re曲线对应的纵坐标的值就是静态介电常数。可以看出,Al、AlNi的静态介电常数分别为58.9、7.95。同时通过观察发现,随着入射光能量的增大,Re曲线的变化趋势大致是先减小后增加至某一值,然后趋于平衡。通过对比分析图6中的非金属C、Si、SiC的实虚部发现,Re曲线随着入射光的能量先增大至某一值后急速减小,最终略微增加至某一值后趋于平衡,而Im曲线先处于平衡状态,当能量增至某一值后,Im曲线急速上升然后减小趋于平衡。
4 结论
本文采用了第一性原理及HSE06交换泛函,对金属Al、Cu、Mg、AlNi、Ti及非金属C、Si、Ge、SiC的结构进行了几何优化,并分析了相应的电子性质及光学性质,得到如下结论:(1)能带结构和电子态密度的分析验证了Cu、Ti、Al、Mg、AlNi的金属性以及C、Si、Ge、SiC的非金属性;同时得出SiC、Ge、Si是半导体,C是绝缘体。(2)晶体结构相同的元素因为电子组态的不同而表现出不同的光学特性。(3)C与Si虽然是同种元素组成的晶体,但由于结构的不同,光学性质也有较大的差异。
