儿童囊性纤维化1例并临床分析
2023-02-14张惠荣
章 伟 袁 博 张 丽 谷 强 张惠荣
石河子大学医学院第一附属医院儿科 (新疆 石河子 832000)
囊性纤维化(CF)是由于囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conduc-tance regulator,CFTR)基因合成、结构及功能异常,使上皮细胞 Cl- 和水分泌减少,导致黏液堆积阻塞呼吸道、胰腺、汗腺等器官的管腔,临床上以汗液氯离子升高、胰腺功能不全及慢性肺部疾病为其主要特点的一种常染色体隐性遗传病[1]。该病在高加索人中常见[2],但在中国罕见[3],2016年被纳入我国罕见病参考名录。目前总结文献发现中国人CF病例CFTR基因突变谱与国外报道大不相同[4],且因CF为一种多向性疾病,临床表现不典型,造成临床诊断困难。现总结我院2021年收治1例初诊为“支气管扩张症、重症肺炎”的患儿,通过详细收集病史资料及进行相关检查,诊断为“囊性纤维化”,报告如下。
1 临床资料及诊治经过
患者,男,13岁,哈萨克族,反复咳嗽、咳痰13年;患者自生后3月起反复出现下呼吸道感染,以咳嗽、咳痰及发热为主要症状,多次诊断为“肺部感染”,抗感染治疗后可好转,但易反复发作,曾被诊断为支气管扩张、支气管哮喘、鼻窦炎、过敏性紫癜及颅内(垂体)占位性病变,已完善检查排除结核。本次于2021年5月初诊为“支气管扩张症、重症肺炎”收住PICU治疗,抗感染治疗好转后为明确支气管扩张及血尿原因转至儿科普通病区。发病以来,患者食欲可,大便次数2-3次/d,常有带油状的稀糊便;患儿父母均为哈萨克族,否认近亲结婚,父母及哥哥均体健。
查体:体温:36.8℃,心率88次/分,呼吸18次/分,血压:120/90mmHg,SPO2 96%(未吸氧),体重35kg,身高:145cm(位于第3百分位),体型消瘦;双副鼻窦无压痛;鸡胸、桶状胸,呼吸平稳,听诊两侧呼吸音对称,双肺呼吸音粗糙,两肺肺底可闻及固定细湿啰音;心腹及神经系统查体未见异常。眼睑、双下肢未见浮肿,未见杵状指(趾)。第二性征未出现。
实验室检查:血常规: WBC 23.9×109/L,N 0.73,L 0.20,Hb 118g/L,PLT 492×109/L,CRP:22.58mg/dl。肝功示:白蛋白:31.9g/L。肾功、心肌酶、血清电解质、血清淀粉酶、尿淀粉酶均未见异常。痰培养及肺泡灌洗液培养均提示铜绿假单胞菌。尿常规提示潜血3+,蛋白(+/-),粪常规提示脂肪球2~5个。胸部HRCT示:双肺支气管扩张合并炎症 (见图1)。电子支气管镜提示气管、支气管内膜炎症,气管、支气管内可见大量粘性分泌物壅塞(见图1)。纤毛活检电镜结果:柱状上皮细胞游离面纤毛大部分缺失,少数细胞表面可见稀疏排列、参差不齐的纤细纤毛,且纤毛发育不良,未见明显微管结构,黏膜下层可见神经丛、血管、线体及散在淋巴细胞浸润(见图2)。基因检测[本部分由北京海思特医学检验公司提供,患方知情同意的前提下,采集患儿及父亲全血各4mL(EDTA)抗凝血)进行,患儿母亲未采集到血清标本],结果见表1:CFTR基因变异,位于7号染色体上chr7:117199646位点出现基因突变,导致c.1521_1523delCTT核苷酸变化,变异类型为纯合。肾穿结果提示:病理诊断:紫癜性肾炎,病变特征:符合局灶性增生性紫癜性肾炎,Ⅲb型。

表1 全外显子测序结果
患者入院后,给与头孢他啶抗感染,雾化吸入,小剂量阿奇霉素(5mg/kg)口服,经电子支气管镜吸痰及肺泡灌洗,并给与营养支持、体位引流,局灶性增生性紫癜性肾炎给与口服泼尼松、吗替麦考酚酯分散片、福辛普利片、左旋咪唑及补充钙剂、维生素D,咳嗽及咳痰减轻。4月后复查肺CT及尿常规较前好转。
2 讨 论
本例患者为哈萨克族儿童,存在慢性肺部疾病,每次经抗感染治疗后均可好转,但病情反复、加重,入院时完善胸部HRCT已提示弥漫性支气管扩张,具有典型的呼吸系统受累临床表现,伴有胰腺功能不全(生长迟滞、脂肪泻、低蛋白血症)表现,基因检测提示CFTR基因存在p.Phe508del纯合突变,因无法进行汗水氯实验,为本例报告一个不足之处,而对该患者完善了水源起皱实验提示时间小于3分钟,经多学科综合团队讨论后诊断为囊性纤维化。
CF为一常染色体隐性遗传的单基因疾病[5],好发于高加索人种,病变累及多个外分泌腺和气管,呼吸系统受累的表现为慢性肺部疾病,因细菌如铜绿假单胞菌、洋葱伯霍尔德杆菌、金黄色葡萄球菌等[6]持续定植于气道而出现慢性的咳嗽、咳痰,肺部影像学常出现支气管扩张、肺浸润影、肺不张,伴有喘息、鼻息肉、慢性鼻窦炎。消化系统受累的表现为小肠性胎粪梗阻、胰腺功能不全以及复发性胰腺炎、营养障碍等。典型的CF病例可表现为汗液氯离子升高、胰腺功能不全及慢性肺部疾病三联征。
囊性纤维化是由CFTR基因突变所致,该基因位于7号染色体长臂3区1带,包含27个外显子,目前已报道 2000多CFTR基因突变位点,仅有10%为常见突变。中国人中最常见的突变为c.2909G>A,p.G970D;c.1766+5G>T;c.3068T>G,p.I1023R[7]。本例行基于家系的全外显子组基因检测发现基因CFTR纯合变异:c.1521_1523delCTT,p.Phe508del(NM_000492),并通过一代测序验证(见图3)。基于ACMG指南该变异可被判定为可能致病(Likely pathogenic),见表2,判断为可能致病的证据包括:PM2 PM4 PP4 PP5。该基因变异导致CF为常染色体隐性遗传,该变异在多个人群数据库中的变异频率极低,符合PM2;该变异为框内缺失变异,符合PM4;另外,该患者表型与该基因型高度吻合,符合PP4;已报道有两例CF病例为该变异所致,并且作者认为变异是致病的,符合PP5。通过一代测序验证发现该变异来自父亲,但患者为纯合变异,父母非近亲结婚,推测母亲携带该变异的可能性很小(母亲未进行一代验证),极有可能该基因为母源性缺失所致。以“p.F508del”“chinese”为关键词在Pubmed中检索,发现2例文献报道,2例患儿均诊断为CF,1例[8]确诊前诊断为支气管扩张,行全外显子组基因检测为p.F508del杂合变异,另1例[9]表现为反复肺部感染、鼻窦炎,同时合并有假性巴特综合征(PBS),行全外显子组基因检测为p.F508del纯合变异。据文献报道[10],虽p.Phe508del是西方国家高加索人中最常见的CFTR变异,约占突变等位基因的70%,该变异在亚洲人群中相对罕见,但本研究提示纯中国人CF患者中也存在纯合子基因型的p.Phe508del。
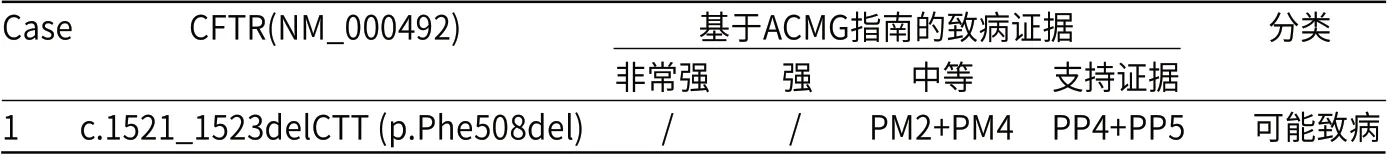
表2 基因变异及基于ACMG指南致病性分析
本例患者病程中出现紫癜性肾炎表现,完善肾穿后明确诊断为局灶性增生性紫癜性肾炎,Ⅲb型。以“Henoch-Schonlein purpura nephritis”“cystic fibrosis”“child”为关键词在Pubmed中检索,未发现文献报道。通过中国知网、万方数据库,以“紫癜性肾炎”“囊性纤维化”“儿童”为关键词检索,未发现文献报告。但Jackson等[11]报道CF患者或敲除CFTR基因的小鼠的肾功能发生了微小的变化,其可能性是在CFTR缺失或者其功能不正常的情况下,CFTR功能被其他转运蛋白所取代。由此推断本例患者的紫癜性肾炎可能与囊性纤维化有关,因为CF是一种多向性疾病,即是指多个不同的,甚至看起来不相关的临床表型由同一个基因控制。但CF患者是否能明确引起紫癜性肾炎及其中的机制有待进一步研究。关于本例CF患儿并发紫癜性肾炎的治疗,因存在铜绿假单胞菌的定植,在口服小剂量的阿奇霉素及加强营养支持的情况下,给与小剂量强的松、吗替麦考酚酯分散片口服治疗,经治疗后复查肺CT及尿常规情况均得到好转。因此,针对CF儿童的治疗,应根据不同的临床表型,提供标准化、个体化的治疗,并且长期全面的随访十分必要[12]。
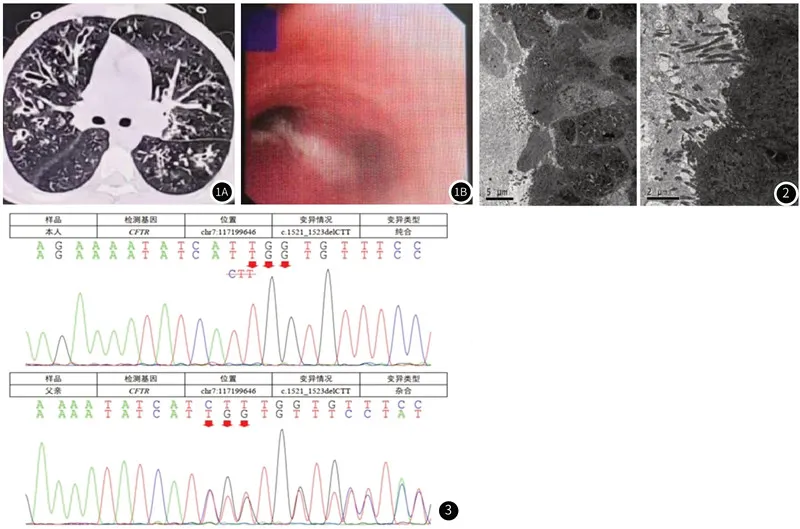
图1 胸部CT提示双肺支气管扩张合并炎症(图1A),电子支气管检查下可见大量粘性分泌物壅塞(图1B)。图2 柱状上皮细胞游离面纤毛大部分缺失,少数细胞表面可见稀疏排列、参差不齐的纤细纤毛,且纤毛发育不良,未见明显微管结构。图3 先证者CFTR基因(NM_000492)c.1521_1523delCTT纯合突变;患者父亲CFTR基因(NM_000492) c.1521_1523delCTT杂合突变。
3 结 论
即使CF仍然是一种罕见的疾病,考虑到中国庞大的人口,CF的发病率存在被低估的可能[13]。因此,儿科医师应提高对该病的认识,对于有慢性肺部疾病病史,或有胰腺功能不全、咸味皮肤的,尤其是高加索人种,应尽早行CFTR基因检测和汗液实验明确诊断。同时本研究发现纯中国人CF患者中存在纯合子基因型的p.Phe508del,并扩大了疾病表型谱。
