多发性骨髓瘤驱动基因的筛选*
2023-02-13苏琪庄文卓李炳宗
苏琪 庄文卓 李炳宗
多发性骨髓瘤(multiple myeloma,MM)是一种恶性增殖的浆细胞肿瘤,其特征是克隆性浆细胞无限增殖,在恶性血液肿瘤发病率中占第二位[1]。近年来,随着蛋白酶体抑制剂、免疫调节剂和CD38 单克隆抗体等药物以及自体干细胞移植越来越多的应用于临床,MM 患者的预后得到极大的改善[2-3]。然而,仍有部分患者并不能从目前的治疗中获益,无法达到有效缓解[4],提示MM 患者的精准分层及个体化治疗是目前亟待解决的问题。
肿瘤是由基因突变驱动的疾病,随着高通量测序技术和癌症基因组学的快速发展,研究人员能够快速分析转录组和基因组的全貌,揭示数目庞大的体细胞突变并找到驱动肿瘤发生发展的关键基因[5]。这些关键基因当以极小的概率发生突变时,将导致恶性克隆扩增。关键基因发生突变将赋予癌细胞生长优势和生存能力,进而驱动肿瘤的发生发展,因此被称为驱动基因[6]。MM 患者的特征之一是由于染色体不稳定性引起的拷贝数和结构变化导致的细胞遗传学异常,这是影响 MM 患者预后的一大重要因素[7]。由此识别潜在的驱动基因将为骨髓瘤患者的个性化靶向治疗提供可能性[8],对于降低MM 的发病率和改善患者的预后将具有重要意义。
在本研究中,首先从84 例患者的含有118 个基因集合的二代测序数据出发,建立1 个标准合理的流程来识别潜藏在MM 患者样本中的驱动基因,并联合来自癌症基因组图谱MM 患者的全基因组测序数据,分析并比较两套数据筛选出的驱动基因及具体突变信息。这些发现将为MM 提供新的重要的基因组改变特征,为后续深入研究MM 的发病机制提供生物信息学依据。
1 材料与方法
1.1 临床资料
回顾性分析2016 年5 月至2020 年11 月在苏州大学附属第二医院诊断并接受治疗的84 例MM 患者初诊时的骨髓单核细胞二代测序结果,采用与MM 发生发展密切相关的118 个基因为靶向测序集合。此外,利用TCGA 数据库(https://portal.gdc.cancer.gov/)下载了205 例MM 患者的全基因组测序数据[9],见图1。本研究通过了苏州大学附属第二医院伦理学审查委员会的批准。
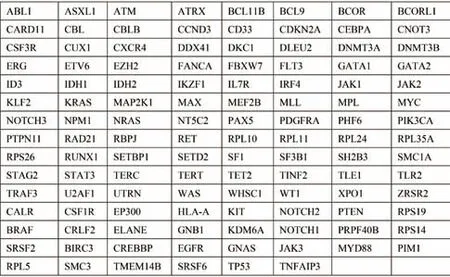
图1 检测的118 个基因列表
1.2 方法
OncodriveCLUST 为一款基于突变聚类的驱动基因识别软件,主要针对功能获得性突变,这些突变通常聚集在蛋白质的特定区域,具有形成突变簇的偏好性,通过对这些突变进行分析来寻找潜在的驱动基因[10]。使用OncodriveCLUST 软件对MM 患者的突变数据进行分析,统计蛋白质上每个位置的功能获得性突变的频率分布并进行聚类,利用每个基因上的簇,对基因进行打分。最后根据P<0.05,错误发现率(false discovery rate,FDR)<0.1 筛选可能的驱动基因。
SomInaClust 是根据突变模式从全外显子组或基因组突变数据中识别候选驱动基因[11]。基本假设是由于选择压力,驱动基因的特征为1)聚类突变;2)肿瘤样本中大量的失活(蛋白质截断)突变。前者是原癌基因(proto-oncogene,OG)的主要模式,后者是抑癌基因(tumor suppressor genes,TSG)的主要模式。随后使用Benjamini &Hochberg 方法对OG 和TSG 的P值进行多次测试校正。根据校正后的P值(qOG 和qTSG),将驱动基因定义为qDG<0.05 的基因。
MutSigCV 主要考虑到肿瘤的异质性,首先根据突变信息建立在肿瘤形成过程中起作用的背景突变模型,根据模型判断每个基因的突变是否比偶然突变频率更高[12]。使用算法软件MutSigCV 对MM 的突变基因进行分析,算出MM 多发性点突变基因发生频率的P值和q值,根据q<0.05 筛选出驱动基因。
1.3 统计学分析
采用R 语言4.1.0 版本对数据进行统计学分析。P值均基于双向假设检验,以P<0.05 为差异具有统计学意义。
2 结果
2.1 驱动基因筛选
首先本研究使用R 中maftools 程序包[13]可视化展示84 例MM 患者的突变信息,如图2A 所示,按突变频率排序展示前40 个基因。本研究采用3 种方法筛选驱动基因。首先,使用OncodriveCLUST 软件识别驱动基因[10],主要针对功能获得性突变进行分析,这些突变通常聚集在蛋白质的特点区域,可能是肿瘤细胞生长优势和肿瘤细胞克隆进化过程中正向选择的信号,通过对这些突变进行分析,来预测潜在的驱动基因。见图2B,NRAS、BCOR、KRAS、TP53 被识别为MM中的驱动基因。其次,使用癌症基因检测软件SomlnaCLUST[11]识别驱动基因,这是一种基于驱动基因在肿瘤样本中的突变模式准确识别驱动基因,并进一步将其分类为癌基因或肿瘤抑制基因的方法。结果显示,NRAS、KRAS、IDH1、CBL 为驱动基因,并被划分为原癌基因(图2C);与其他研究结果相一致的是TP53具有较高的原癌基因分数[14-15]。最后,本研究应用基于频率的MutSigCV 进行突变负荷分析寻找MM 的驱动基因[12]。驱动基因信息如表1 所示,以q<0.05 为阈值,NRAS、KRAS、DNMT3A、TP53 被识别为驱动基因。
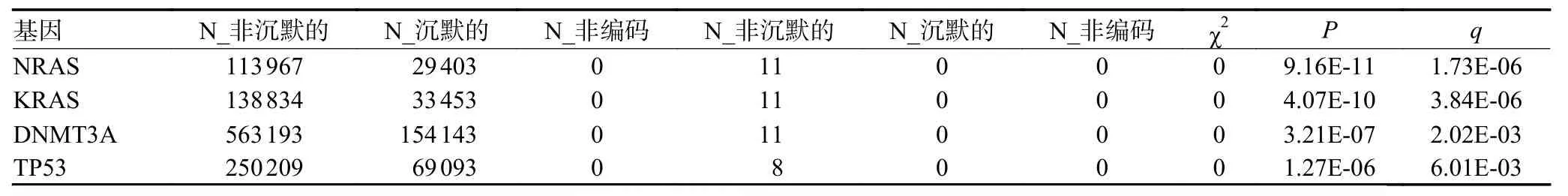
表1 Mutsigcv 识别驱动基因
由于本研究所提供的84 个MM 组织样本的二代测序集合仅包含与MM 发生发展密切相关的118 个基因,虽节省人力、物力、财力及时间,但识别出的驱动基因有限。因此为了验证此种方法的可靠性,本研究从癌症基因图谱(TCGA,https://cancergenome.nih.gov/)获取了205 例MM 患者样本的全基因组测序数据[9],进行驱动基因识别并比较两套数据的基因突变信息。
如图2 所示,首先使用maftools 将205 例MM患者的突变信息进行可视化展示,结果显示KRAS、NRAS 等基因的突变频率较高(图3A),这与靶向突变测序结果一致。接下来使用OncodriveCLUST、SomlnaCLUST、MutSigCV 软件分别寻找驱动基因。OncodriveCLUST 识别出NRAS、KRAS、IRF4、BRAF和ACTG1 为驱动基因(图3B)。SomlnaCLUST 识别出NRAS、KRAS、BRAF、IDH1 为原癌基因,TP53同样具有较高的原癌基因分数(图3C)。KRAS、NRAS、TP53、DIS3、ZNF717、BRAF、TRAF3 被MutSigCV 识别为驱动基因(表2)。分析结果可知,NRAS、KRAS、TP53、IDH1 为两套数据共同识别出的驱动基因。
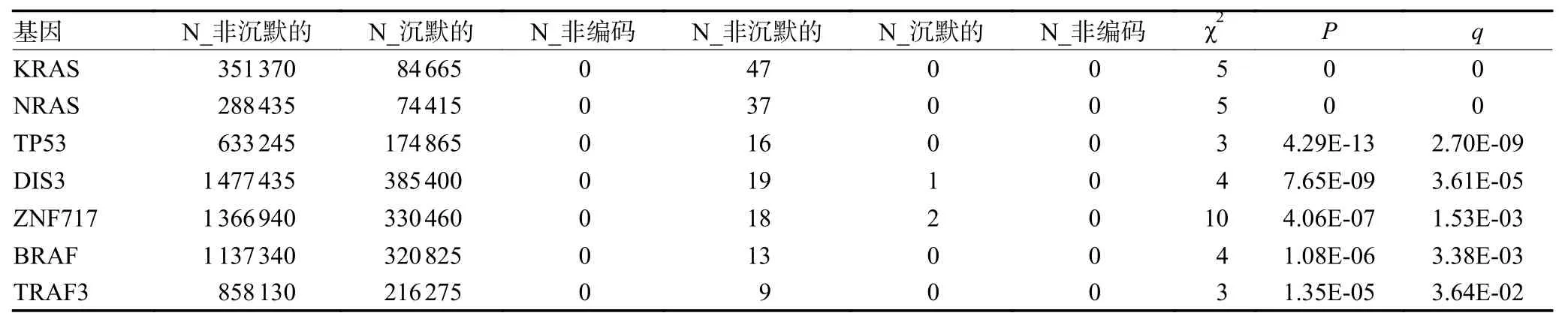
表2 Mutsigcv 识别驱动基因
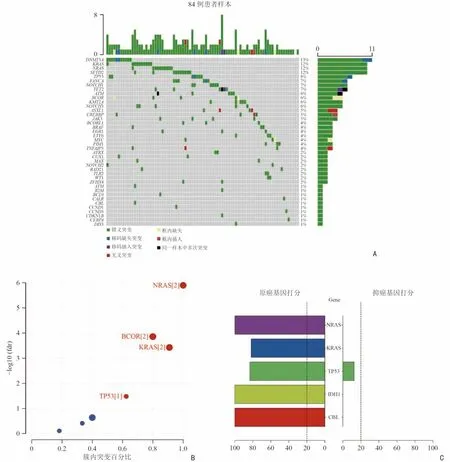
图2 驱动基因筛选结果
2.2 两套数据基因突变信息的比较
为更好地比较两套MM 患者的基因突变情况,本研究采用coOncoplot 函数接受两个对象并将它们并排绘制成条形图。结果如图3 所示,两套数据均显示,KRAS、NRAS 和TP53 的突变频率高。本院84 例MM 患者的靶向测序结果中,DNMT3A 突变频率最高(图4A)。随后将突变基因进行致癌信号通路的富集,结果显示两套MM 患者的突变基因在NOTCH、RTK-RAS、Cell-Cycle、TP53、MYC 及WNT 通路均有富集(图4B)。许多致癌基因具有比其他基因座更频繁突变的优先位点,这些位点被认为是突变热点,棒棒糖图可以用于显示突变热点以及其他突变位点。利用Lollipopchart 将单个基因的突变位点及其所在蛋白结构域进行可视化展示,并比较两套患者基因突变数据中显著突变的NRAS、KRAS、TP53 和IDH1。结果如图4C 所示,两套数据中高频突变的基因其发生突变的位点一致。
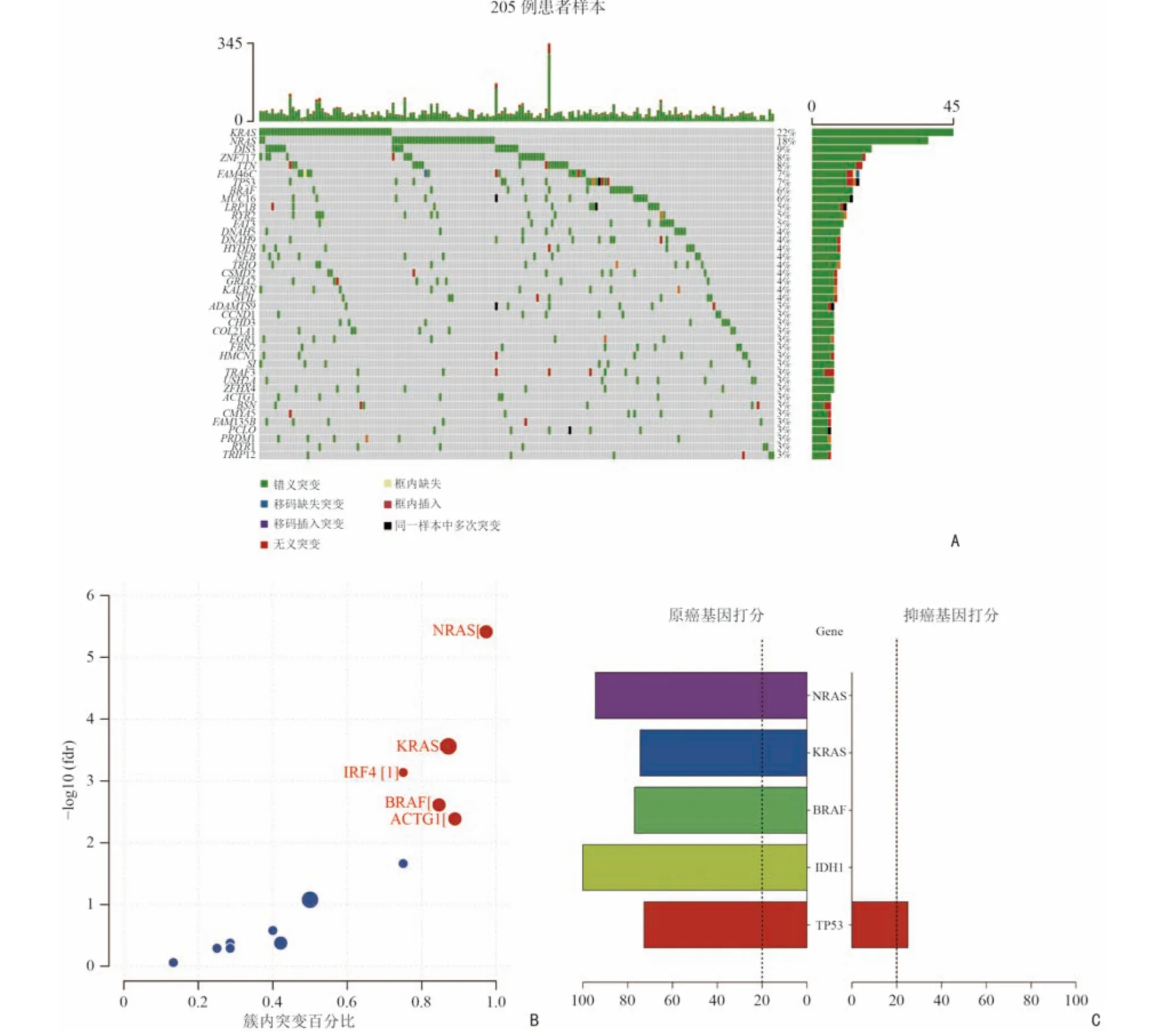
图3 驱动基因筛选结果
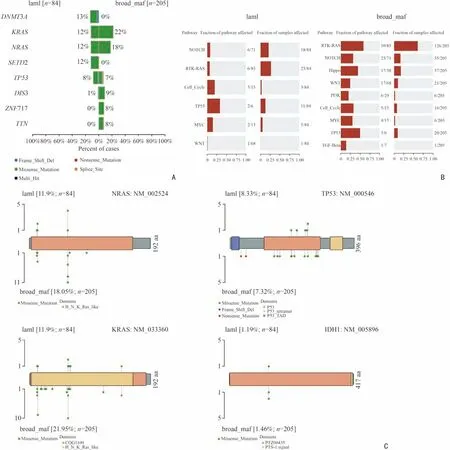
图4 两套数据基因突变信息的比较
2.3 基因突变之间共同发生和相互排斥的模式
为观察两套数据中基因突变的模式,本研究采用somaticInterations 绘制基因间相互排斥或共发生事件的情况。如图5A 所示,在84 例MM 患者靶向测序数据中,以P<0.05 为阈值,本研究发现突变基因中NOTCH1 与ATRX、JAK3;FANCA 与SETD2 存在显著共发生模式。而在205 例MM 患者突变基因中存在共发生与相互排斥的基因明显较多(图5B)。这可能与样本的统计量及基因的统计量有关。
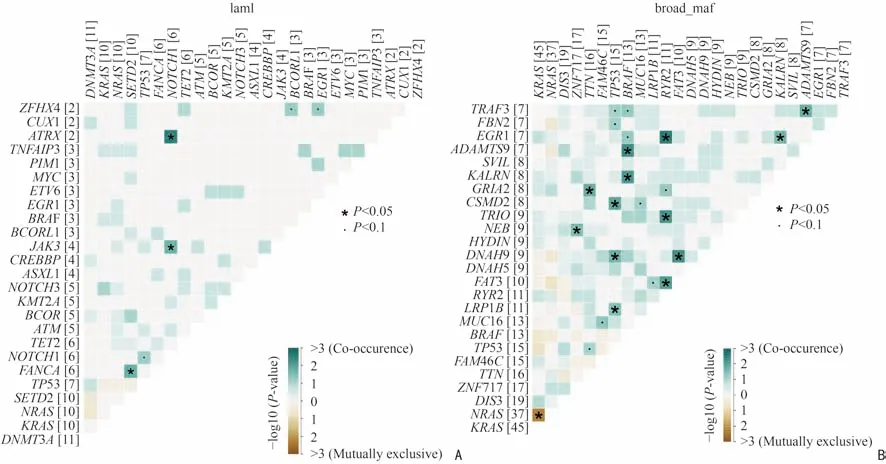
图5 基因之间的突变模式
2.4 驱动基因的鉴定
通过比较来自本院的84 例和来自TCGA 数据库的205 例MM 患者突变数据的驱动基因分析结果,研究发现CBL、BCOR 和DNMT3A 是新鉴定出的驱动基因,并将其突变位点绘制如下(图6A)。已有研究报道DNA 甲基转移酶DNMT3A(DNA methyltransferase 3 alpha,DNMT3A)通过调控DAB2IP 介导的MEK/ERK 激活促进结直肠癌进展[16]、且DNMT3A在急性髓系白血病(acute myeloid leukemia,AML)患者中具有较高突变频率,并且与不良预后密切相关[17]。此外,原癌基因CBL(Cbl proto-oncogene,CBL)的突变通过增加与LYN 和PIK3R1 的相互作用驱动致癌信号通路PI3K/AKT[18]。BCL6 辅抑制因子(BCL6 corepressor)BCOR 最近也被证明其突变在成熟T 细胞淋巴瘤中具有高复发性和致癌性[19]。此外,本研究针对CBL、BCOR 和DNMT3A 3 个新鉴定的驱动基因在两套数据中的差异表现进一步分析。结果显示在205 例MM 患者中,CBL、DNMT3A 无突变,BCOR 中仅1 例突变(图6B)。随后将BCOR 在两套患者基因突变数据中的突变位点通过棒棒糖图可视化 展示并比较,结果如图6C 所示,两套患者基因突变数据中BCOR 的突变位点无重合,由此推测本选取的差异和样本统计量有限。

图6 新型驱动基因的突变信息
3 讨论
MM 是一种分子水平异质性很大的恶性肿瘤,在精准医疗模式下,分子分型的差异在很大程度上决定了患者的个体化治疗方案[20-21]。目前,对于MM 的分子机制的研究越来越多,但由于其调控网络的复杂性,需要对MM 发生发展过程中的关键驱动因子做更深入的挖掘,寻找特异性更高的标志物,以期为骨髓瘤患者提供更好的治疗方案。
癌症驱动因素的识别是解释癌症发生机制和实现精准医疗的关键挑战。根据单个突变位点或整个基因来识别癌症驱动因素的方法有很多,但各具优缺点。MutSigCV 基于基因突变的频率,通过识别基因与背景突变的关系来定义驱动基因。背景突变模型用于量化乘客突变的积累,过高或过低的背景突变率的估计会导致结果不准确。同义突变常被用作固定的背景模型。然而,研究表明背景突变并不是均匀分布的。因此,准确估计背景突变率仍然具有挑战性。Oncodrive-CLUST,一种识别蛋白质序列中具有显著突变聚类倾向的基因的方法。该方法通过编码沉默突变构建背景模型,将背景率阈值以上多个突变的位置识别为潜在有意义的簇。该方法识别出具有功能获得性突变的癌基因,但在预测以功能丧失性突变为特征的肿瘤抑制基因方面不足。然而,以上两种方法通常不能检测不太频繁的突变基因,也不能区分原癌基因或肿瘤抑制基因。SomInaClust 是一种基于驱动基因在肿瘤样本中的突变模式来准确识别驱动基因,并进一步将其分类为癌基因或肿瘤抑制基因的方法。该方法使用参考突变数据:1)确定突变热点,即蛋白编码区(coding sequences,CDS )位置在研究样本中包含比偶然预期更多的突变;2)计算基因背景突变率。SomInaClust 是对其他候选驱动基因识别方法的补充。
从比较的结果看出,MutSigCV、OncodriveCLUST、SomInaClust 可以互相发现一些其他检测方法忽略的基因。因此,在检测肿瘤的驱动基因时,除去样本量是影响算法性能的稳定性外,建议使用基于不同原理的软件综合检测。
在本研究中,首先以回顾性分析的本院初诊的84 例MM 患者的118 个基因的靶向二代测序数据为参考数据,使用基于突变聚类、突变模式及突变频率的算法识别潜在的驱动基因。而后分别将上述寻找驱动基因的算法应用于TCGA 数据中。通过分析并比较两套体细胞突变数据筛选出的驱动基因,结果显示在84 例使用靶向测序集合的体细胞突变数据中驱动基因:NRAS、BCOR、KRAS、TP53、IDH1、CBL、DNMT3A;在205 例使用全基因组测序的TCGA 体细胞突变数据中驱动基因:IRF4、ACTG1、BRAF、
IDH1、KRAS、NRAS、TP53、DIS3、ZNF717、TRAF3。本研究发现NRAS、KRAS、TP53、IDH1 为两套数据共同识别出的驱动基因。随后,通过比较两套数据中具体的突变信息,本研究观察到KRAS、NRAS 和TP53 的突变频率显著较高。通路富集分析发现两套MM 患者的突变基因在NOTCH、RTK-RAS、Cell-Cycle、TP53、MYC 及WNT 通路均有富集。比较两套患者基因突变数据中高频突变的NRAS、KRAS、TP53 和IDH1,发现在两套MM 数据中发生突变的位点一致。此外,通过比较来自本院的84 例和来自TCGA 数据库的205 例MM 患者突变数据的驱动基因分析结果,本研究找出CBL、BCOR 和DNMT3A为新鉴定的驱动基因。
综上所述,此研究通过多数据整合的方法,鉴定了MM 的潜在驱动基因,为MM 提供新的重要的基因组改变特征。本研究所鉴定出的驱动基因可能为今后进一步通过科研实验手段探索MM 发病机制提供新的线索。
