第二类创新医疗器械审批现状及监管的几点思考
2023-02-12段世梅仲志真
【作 者】 段世梅,仲志真
上海市医疗器械化妆品审评核查中心,上海市,200020
0 引言
医疗器械作为国家现代化医疗卫生体系建设中的基础装备,直接关系到人民群众的生命安全和健康福祉。在过去的几十年里,我国逐渐跻身为医疗器械进口、制造、出口大国。中国医疗卫生事业和庞大人口的医疗保健服务,不可能完全依赖进口产品,尤其在重大公共卫生危机状态下,医疗器械具有显著的 卡脖子效应,高端进口医疗器械市场长期被跨国公司垄断,国产医疗器械竞争力薄弱,人民群众医疗费用居高不下的问题仍待解决。为了摆脱这种状况,近年我国愈发重视医疗器械行业创新[1]。特别在高端医疗器械领域提出了国产替代,并发布了创新医疗器械审批程序[2],医疗器械的自主创新及相关产品的国产化,对国民健康保障具有极其重要的意义。
1 第二类创新医疗器械审批现状
2015年后,广东、浙江、山东、江苏、四川、江西、陕西、湖南、河南等地分别出台了第二类创新医疗器械特别审批程序,截至目前,各省市拟进入第二类创新医疗器械公示数量如图1所示(数据来源于各省市局网站公示)。
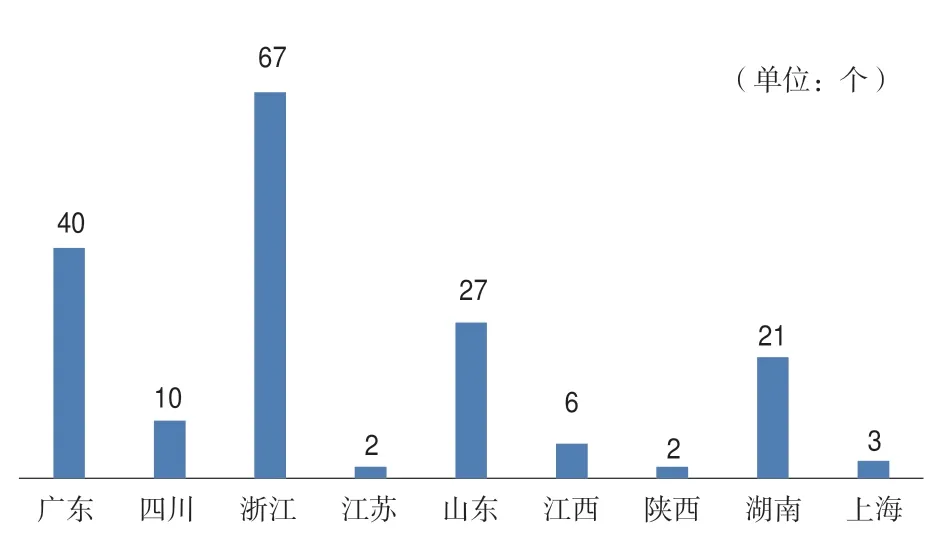
图1 各省市第二类创新医疗器械公示数量Fig.1 Number of class II innovative medical devices publicized by provinces and cities
选取广东、浙江、山东、湖南为例对进入创新医疗器械公示产品进行类型分布分析,由表1可以看出,有源医疗器械在第二类创新医疗器械中的占比远高于无源医疗器械和体外诊断试剂。通过对上海第二类创新医疗器械的申报情况梳理,有源医疗器械(包括医用软件)在第二类创新医疗器械申报产品中占比为54.2%,与其他省市情况类似,占比较高。有源医疗器械申报量的大幅上涨,主要是数字化转型带来的改变。随着大数据、人工智能及穿戴式设备的发展,未来该类产品的创新比例可能会进一步上升。
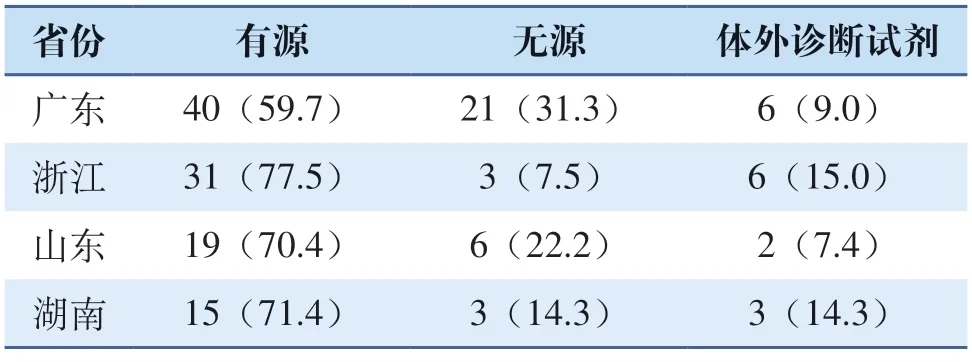
表1 选取省份产品类型分布情况表 [n(%)]Tab.1 Distribution of product types in selected provinces
2020年2月,上海市药品监督管理局对外发布了《上海市第二类创新医疗器械特别审查程序》[3],为第二类创新医疗器械开辟了专用通道,企业创新积极性高涨,产业迅速发展。目前第二类创新医疗器械的来源主要有临床实践创新、新技术在医疗器械上的应用及已有医疗器械迭代新技术后的换代产品等3个方面。
自2020年2月至今,共计收到第二类创新医疗器械审批申请24件,其中有源医疗器械13件,占比54.2%;无源医疗器械10件,占比41.6%;体外诊断试剂1件,占比4.2%。24件中符合形式审查要求予以受理15件,不予受理9件,主要原因是申报资料不齐全和申报产品管理类别存疑。
受理的第二类创新医疗器械,目前仅有3件(2件有源医疗器械,1件无源医疗器械)第二类创新医疗器械产品获批进入创新医疗器械通道。通过分析本市的第二类创新医疗器械审查情况,发现通过率比较低的原因主要有以下几点:
(1)产品主要的作用原理/作用机理经审查后属于非国内首创产品。
(2)产品的性能或安全性有改进,但无根本改进,不能判断技术上是否处于国际领先水平。
(3)申报产品提交的发明专利不是产品的核心技术或提交的核心发明专利与申报产品无明显关联性。
(4)申报创新医疗器械与已在临床应用的同类产品相比,或者与临床的 金标准 相比,无显著的临床应价值,或通过引进国外成熟产品进行升级,但未提交升级后相关临床前研究和临床试验资料,缺乏证明申报产品具有显著临床应用价值的证据。
综上,不予通过创新医疗器械审批一方面是因为注册申请人申报产品本身不符合该程序规定的相关要求;另一方面是因为注册申请人提交的申报材料不足以证明其满足该程序的要求。
2 创新研究成果转化为医疗器械上市的难点
大量现代科学技术前沿成果的汇集,使医疗器械创新产品不断涌现,但创新研究成果在转化为产品时仍遇到不少难点,一定程度上导致创新医疗器械上市进程缓慢。
2.1 产品技术要求制定环节面临的难点
对于创新医疗器械,由于没有公认的评价思路和方法可参考,也没有相应的国家标准或行业标准,注册申请人需通过研发设计确定产品的性能指标,导致制订产品技术要求、性能指标和确定检验方法周期过长,如在设计开发时未考虑部分安全风险点,审评环节可能面临补充检测,会进一步导致上市进程缓慢。
2.2 临床评价环节面临的难点
对于创新医疗器械,由于其在结构与工作原理、材料构成与特性、软件算法等方面与已获批上市医疗器械存在明显差异,有些创新医疗器械甚至开创了某一领域的全新治疗方向,按照《医疗器械临床评价技术指导原则》的规定,只能通过临床试验路径进行临床评价,导致成本上升、产品上市进程延长。
2.3 质量管理体系面临的难点
医疗器械注册质量管理体系,决定了产品是否能够注册上市,也承担着保证上市产品真实安全和稳定可靠。创新医疗器械的注册申请人一般为初创企业、医疗机构、科研机构,重视样品实物,忽略质量管理体系的建立,缺少专业的质量体系管理人员,导致不能提供样品在质量管理体系下运行的相关记录,可能会在注册核查时无法提供注册检验用样品的生产记录和检验记录,导致无法得出申请产品样品的真实性结论。
3 第二类创新医疗器械监管的几点思考
依据本市以往第二类创新医疗器械的审批经验,针对创新医疗器械审批建议可通过建立健全制度规章,转变审评审批思路,将服务延伸至创新医疗器械产品研发及临床试验方案制订等环节[4],进一步推进创新医疗器械上市进程,促进科研成果的快速转化。
(1)提前布局,提高能力。提高创新医疗器械提前服务能力,通过预测未来几年具有战略性、前瞻性的医疗器械关键技术点,设立科研项目;同时引导和鼓励有条件的科研院所、高等院校和临床医院的专家,积极与创新企业开展合作,共同探索产品的技术要求和检测方法制订,以期提高申报资料的质量。
(2)强化绿色通道,落实提前介入服务,加大技术审评与注册核查的同步机制。对进入创新审批程序的医疗器械指定专人负责对接,开展提前介入服务,如对注册申报资料开展审查,同时积极开展上门调研、法规宣贯、办理流程介绍、生产质量管理体系指导、企业问题咨询解答等服务,助力企业更好地了解法规、流程,提前做好准备,少走弯路,缩减审评核查耗时。
(3)拓展咨询服务渠道,优化现场咨询机制。以临床需求为导向,对医疗机构为主导研发的创新医疗器械,积极参与产品技术要求的制订研究工作中的答疑解惑,深入医疗机构探讨医疗器械创新产品的研发和验证工作,切实帮助临床机构解决研发中遇到的困难和问题。主动联系各创新医疗器械服务站和相关园区,提前了解拟申报创新医疗器械企业的各种需求,对企业开展相关业务指导,侧面提高申报资料的质量。
(4)加强创新医疗器械事中事后监管制度衔接。依据创新医疗器械的特点,探索定期提交创新医疗器械客户投诉记录、年度不良事件报告、临床使用情况报告及召回情况报告;监管部门适时以数据库的方式公布创新医疗器械使用的相关信息。由于创新医疗器械在使用上也具有独特性和创新性,为了做好使用环节的监管,建议制定创新医疗器械使用质量监督管理办法,规范创新医疗器械的使用。
监管部门可通过不定期走访企业,了解企业存在的问题及困惑,定期总结,适时调整监管政策。随着医疗器械监管政策的不断完善,创新医药器械未来的方向将会以高端医疗设备国产化、高端医疗设备的短板为重心,突破技术装备瓶颈,实现高端医疗器械的自主可控,也为人民群众的生命安全和健康造福。
