新型手性硫醚-亚磷酰胺配体的设计与应用
2023-02-06林舒妤彭丽燕张雪莲游歌云
冯 彬, 林 瑶, 林舒妤, 彭丽燕, 张雪莲, 游歌云
(百色学院 化学与环境工程学院 桂西区域污染与治理广西高校重点实验室,广西 百色 533000)
光学活性化合物在医药、农药、材料及生命科学领域中具有重要的应用潜力。过渡金属催化的不对称合成方案具有高选择性和高活性等特点,成为合成光学活性化合物研究热点之一[1-3]。自1966年Noyori小组报道了第一例手性过渡金属配合物催化的不对称反应以来,手性配体的开发一直是过渡金属催化的重要领域[4-6]。部分配体因其催化反应多样,底物适用范围广而被称为是具有“幸运结构”的手性配体[7-8]。
手性硫醚配体结构相对稳定而且与过渡金属配位能力强,配位后会在硫原子处产生一个新的手性中心,能够为中心金属提供额外的手性环境,其开发与应用成为近年来有机化学家们的研究热点。一系列手性双硫配体、手性硫膦配体、手性硫氮配体和手性硫氧配体等在被不断开发报道[9-12]。得益于金属-硫醚络合物的种类多样,其在金属催化的手性合成中的应用也越来越广泛,如烯丙基化反应、偶极环加成反应和不对称转移氢化反应[13-15]。
与此同时,亚磷酰胺结构配体具有易于修饰、结构多样和反应适用范围广的特点,手性亚磷酰胺与其他配位原子组合的配体设计与应用逐渐增多[16]。例如2011年DU课题组报道了亚磷酰胺-烯烃配体,该配体在钯催化的吲哚烯丙基化和去芳化烯丙基化反应中具有非常优异的立体控制效果。通过对比发现,配体中的双键对反应的顺利发生具有重要影响[17-18]。2003年,DOHERTY报道了8-氨基喹啉衍生的亚磷酰胺配体在铑催化的烯烃不对称氢化中的应用,在最佳条件下,原料能够以95%的对映选择性转化为产物[19]。BAKOS报道的具有柔性碳链结构的亚磷酰胺配体以铑做催化剂,在环境友好性的碳酸酯溶剂中能够以99.6%的对映选择性得到氨基酸类化合物[20]。HU课题组报道的亚磷酰胺-膦配体在亚胺的不对称氢化中能够以最高99%的对映选择性得到手性胺类化合物[21]。CREVISY和MAUDUIT课题组报道了硫醚衍生的亚磷酰胺配体,其铜/配体络合物能够以中等到优秀的对映选择性催化二乙基锌与不饱和酮的亲核加成反应[22]。近期,XIAO课题组报道了一系列具有硫醚亚磷酰胺结构的硫醚配体,其在过渡金属催化及路易斯酸催化中均具有非常优异的立体控制效果[23-25]。
上述研究表明,在亚磷酰胺的基础上通过骨架的合理组合引入配位原子后所构建的新配体往往能够表现出优异的催化效果和立体控制。鉴于此,为了探究手性硫醚配体更加广泛的应用,本课题组设计并合成了一种结构多样及合成步骤简单的硫醚-手性亚磷酰胺配体。以2-氟苯甲醛1、硫酚(醇)2和手性R-(α)-甲基苄胺5为原料将各个片段进行逐步拼接得到胺基硫醚4,最后利用模块组装的手性配体构建方法,得到了同时具有轴手性和中心手性的硫醚-亚磷酰胺配体(图1)。该方法能够快速构建手性膦-硫配体库,为后续的反应开发提供强有力的支撑。由于钯催化的不对称烯丙基化反应是构建手性碳碳键和碳杂原子键的重要合成方法,同时钯催化的丙二酸二甲酯和1,3-二苯基烯丙基醋酸酯的烯丙基化反应也是评价配体催化活性和立体控制效果的经典反应。因此,在得到了结构多样的配体后,随即以钯催化的丙二酸二甲酯和1,3-二苯基烯丙基醋酸酯的烯丙基化反应作为模板反应,探究了配体在该反应中的活性[26-27]。

图1 代表性的具有亚磷酰胺结构的手性双齿配体
1 实验部分
1.1 仪器与试剂
X-4型显微熔点测定仪(郑州杜甫仪器厂);SGW-3型自动旋光仪(上海仪电物理光学仪器有限公司);Bruker 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标,德国布鲁克公司);Q-TOF6550型四杆质谱仪(美国安捷伦科技有限公司);Bruker micrOTOFII ESI-TOF型质谱仪(德国布鲁克公司);Infinity 1200型高效液相色谱仪(流动相为正己烷和异丙醇,美国安捷伦科技有限公司);Chiralpak AD-H型手性色谱柱[大赛璐药物手性技术(上海)有限公司]。
乙酸乙酯和石油醚均购自上海泰坦科技有限公司,所用溶剂均为超干溶剂,其余所用试剂均为分析纯或化学纯,购自上海泰坦科技有限公司或上海迈瑞尔公司。
1.2 合成
(1) 2-硫醚取代芳醛的合成[28]
在室温下,将2-氟苯甲醛(50.0 mmol)、碳酸钾(100.0 mmol)与硫酚(醇)(50.0 mmol)溶解于二甲亚砜(DMSO)50 mL中,加热至100 ℃,反应至终点。抽滤,滤饼用乙酸乙酯(3×20 mL)洗涤,滤液与洗液合并转入分液漏斗分液,用饱和氯化铵溶液100 mL洗涤,收集有机相,水相用乙酸乙酯(3×20 mL)洗涤,合并有机相,用无水硫酸钠干燥,抽滤,滤液拌硅胶粉减压脱去溶剂,经硅胶柱层析(洗脱剂:石油醚∶乙酸乙酯=50∶1,V∶V)纯化,减压脱去层析液得中间体3a~3g:
3a: 淡黄色固体,收率85%;1H NMR(400 MHz, CDCl3)δ: 10.34(s, 1H), 7.83(s, 1H), 7.34(s, 3H), 7.31~7.23(m, 1H), 7.23~7.16(m, 2H), 7.06~6.93(m, 1H), 2.37(s, 3H);13C NMR(100 MHz, CDCl3)δ: 191.48, 142.80, 138.98, 134.01, 133.88, 132.99, 132.27, 130.52, 129.08, 128.74, 125.54, 21.19。
3b: 淡黄色固体,收率90%;1H NMR(400 MHz, CDCl3)δ: 10.33(s, 1H), 7.90~7.80(m, 1H), 7.44~7.37(m, 1H), 7.36~7.29(m, 5H), 7.05(dd,J=7.9 Hz, 1.2 Hz, 1H);13C NMR(100 MHz, CDCl3)δ: 191.42, 140.96, 134.74, 134.50, 134.11, 133.51, 132.35, 131.60, 129.94, 129.88, 126.38。
3c: 淡黄色固体,收率81%;1H NMR(400 MHz, CDCl3)δ: 10.42(s, 1H), 7.87(dd,J=7.6 Hz, 1.7 Hz, 1H), 7.48~7.26(m, 4H), 7.02~6.82(m, 2H), 3.82(s, 3H);13C NMR(100 MHz, CDCl3)δ: 191.73, 158.66, 140.61, 134.53, 133.92, 133.86, 131.56, 130.29, 130.27, 126.13, 121.47, 120.92, 111.21, 55.78。
3d: 淡黄色固体,收率81%;1H NMR(400 MHz, CDCl3)δ: 10.33(s, 1H), 7.85(s, 1H), 7.56~7.10(m, 6H), 6.83(s, 1H), 2.36(s, 3H);13C NMR(100 MHz, CDCl3)δ: 191.42, 141.78, 141.62, 135.14, 133.96, 132.94, 132.67, 131.18, 130.92, 129.31, 128.25, 127.20, 125.38, 20.49。
3e: 淡黄色固体,收率86%;1H NMR(400 MHz, CDCl3)δ: 10.33(s, 1H), 7.81(d,J=7.5 Hz, 1H), 7.30~7.15(m, 5H), 6.54(d,J=7.9 Hz, 1H), 2.38(s, 6H);13C NMR(100 MHz, CDCl3)δ: 191.28, 144.03, 142.70, 133.91, 133.64, 132.15, 129.79, 129.21, 128.66, 125.17, 124.20, 21.57。
3f: 白色晶体,收率77%;1H NMR(400 MHz, CDCl3)δ: 10.24(s, 1H), 7.80(d,J=7.7 Hz, 1H), 7.51~7.41(m, 2H), 7.34~7.20(m, 6H), 4.12(s, 2H);13C NMR(100 MHz, CDCl3)δ: 191.50, 140.97, 136.06, 134.52, 133.93, 131.63, 129.74, 128.86, 128.58, 127.46, 126.04, 38.77。
3g: 淡黄色液体,收率88%;1H NMR(400 MHz, CDCl3)δ: 10.80(s, 1H), 8.00(d,J=7.6 Hz, 1H), 7.66~7.49(m, 3H), 1.30(s, 9H);13C NMR(100 MHz, CDCl3)δ: 193.66, 139.96, 139.41, 136.59, 133.54, 129.52, 128.06, 47.50, 30.87。
(2) 手性胺基硫醚的合成[25]
将中间体醛3(10.0 mmol)和R-(α)-甲基苄胺5(12.0 mmol)溶解在甲醇100 mL中,搅拌直至醛完全消失,降至0 ℃,再将硼氢化钠(20.0 mmol)分批加入到反应体系中搅拌,反应至终点。减压脱去甲醇,然后加入饱和氯化铵100 mL,用二氯甲烷(3×20 mL)萃取,合并有机相并用无水硫酸钠干燥,抽滤,滤液拌硅胶粉在减压下脱去溶剂,经硅胶柱层析(洗脱剂:石油醚∶乙酸乙酯=20∶1,V∶V)纯化,减压脱去层析液后得到二级胺中间体4a~4g:
4a: 无色液体,收率79%;1H NMR(400 MHz, CDCl3)δ: 7.38~7.12(m, 11H), 7.08(d,J=8.0 Hz, 2H), 3.84~3.62(m, 3H), 2.31(s, 3H), 1.29(d,J=6.6 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 145.38, 140.80, 136.93, 134.93, 132.42, 131.98, 130.88, 130.01, 129.99, 128.35, 127.75, 127.27, 126.83, 126.75, 57.45, 50.13, 24.48, 21.04。
4b: 无色液体,收率82%;1H NMR(400 MHz, CDCl3)δ: 7.47~7.16(m, 11H), 7.08~6.91(m, 2H), 3.80~3.64(m, 3H), 1.29(d,J=6.6 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 136.15, 134.36, 132.75, 132.14, 130.68, 130.35, 128.58, 128.44, 128.13, 127.00, 126.76, 120.18, 57.62, 49.99, 24.27。
4c: 无色液体,收率86%;1H NMR(400 MHz, CDCl3)δ: 7.37~7.27(m, 7H), 7.26(s, 1H), 7.25~7.15(m, 3H), 6.95~6.71(m, 3H), 3.86(s, 3H), 3.82~3.67(m, 3H), 1.27(d,J=6.6 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 156.71, 145.47, 142.47, 134.28, 132.40, 130.25, 129.95, 128.35, 128.10, 127.94, 127.61, 126.81, 126.79, 124.94, 121.26, 110.69, 57.47, 55.83, 50.18, 24.49。
4d: 无色液体,收率81%;1H NMR(400 MHz, CDCl3)δ: 7.30~7.19(m, 5H), 7.18~7.05(m, 3H), 7.10~7.02(m, 3H), 7.02~6.92(m, 2H), 3.81~3.51(m, 3H), 2.26(s, 3H), 1.21(d,J=6.5 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 145.37, 140.96, 138.54, 134.69, 133.91, 132.17, 130.93, 130.45, 130.08, 128.36, 127.87, 127.30, 127.07, 126.84, 126.73, 126.68, 77.32, 77.00, 76.68, 57.44, 50.07, 24.45, 20.41。
4e: 白色固体,收率90%;1H NMR(400 MHz, CDCl3)δ: 7.48~7.32(m, 4H), 7.30~7.13(m, 6H), 7.07~6.95(m, 2H), 6.44(d,J=7.8 Hz, 1H), 3.90~3.75(m, 3H), 2.37(s, 6H), 1.41(d,J=6.6 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 145.50, 143.85, 136.94, 136.79, 130.39, 129.32, 129.19, 128.46, 128.38, 127.57, 126.87, 126.80, 124.89, 124.50, 57.40, 49.83, 24.55, 21.75。
4f: 无色液体,收率78%;1H NMR(400 MHz, CDCl3)δ: 7.42~7.28(m, 6H), 7.27~7.10(m, 9H), 4.05(d,J=13.6 Hz, 2H), 3.78~3.56(m, 3H), 1.31(d,J=6.6 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 137.32, 135.20, 130.63, 129.64, 128.80, 128.45, 128.37, 127.60, 127.19, 126.88, 126.82, 126.59, 57.59, 49.91, 39.33, 24.44。
4g: 无色液体,收率84%;1H NMR(400 MHz, CDCl3)δ: 7.60(d,J=7.6 Hz, 1H), 7.43~7.12(m, 9H), 3.88(d,J=13.0 Hz, 1H), 3.80~3.69(m, 2H), 1.35(d,J=6.6 Hz, 3H), 1.23(s, 9H);13C NMR(100 MHz, CDCl3)δ: 139.00, 131.91, 130.14, 129.01, 128.39, 127.02, 126.93, 57.45, 50.64, 47.31, 31.05, 24.38。
(3) 手性硫醚-亚磷酰胺配体的制备[25]
在0 ℃和氮气保护条件下,将二异丙基乙胺(30.0 mmol)和三氯化磷(6.0 mmol, 3.0 mL, 2.0 M 二氯甲烷溶液)溶解在二氯甲烷20 mL中,手性胺基硒醚中间体(5.0 mmol)溶于二氯甲烷5mL中,逐滴加入上述体系,在室温下反应4 h, (R)-联萘二酚((R)-BINOL, 7.5 mmol)溶于二氯甲烷25 mL中,0 ℃条件下逐滴加入体系。TLC检测反应,反应完成后拌硅胶粉减压下脱去溶剂,经硅胶柱层析(洗脱剂:石油醚∶乙酸乙酯=30∶1,V∶V)纯化,即得到目标配体L1~L9(图2)。

图2 手性硫醚-亚磷酰胺配体

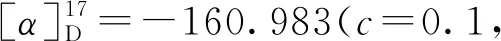







(4) 烯丙基烷基化反应产物的制备
在氮气保护下,烯丙基氯化钯二聚体0.01 mmol和配体0.044 mmol溶于二氯甲烷2.0 mL中并在室温下搅拌1 h,再向体系中加入烯丙基醋酸酯0.2 mmol、碳酸铯0.6 mmol,升温至40 ℃搅拌10 min,丙二酸二甲酯0.3 mmol加入体系反应,搅拌直至反应完全。加入饱和氯化铵溶液10.0 mL终止反应,用二氯甲烷(3×5.0 mL)萃取,有机相合并后无水硫酸钠干燥。干燥后的有机相抽滤,滤液拌硅胶粉减压脱去溶剂,经硅胶柱层析(洗脱剂:石油醚∶乙酸乙酯=5∶1,V∶V)纯化,减压脱去层析液即得烯丙基烷基化产物9。

2 结果与讨论
2.1 溶剂对烯丙基化反应的影响
以L2作为模板配体、rac-1,3-二苯基烯丙基醋酸酯7和丙二酸二甲酯8作为原料,探究了溶剂对钯催化的不对称烯丙基化反应的影响(表1)。该反应体系无论使用卤素类溶剂、醚类溶剂或是配位性的乙腈反应均能在较短时间内反应完成,而使用芳香性溶剂均三甲苯时,反应时间大幅度延长,二氯甲烷能够给出最好的结果。通过与文献所报道结果(HPLC保留时间和比旋光度)做对比,产物的绝对构型为R[20]。

表1 反应溶剂的优化
2.2 配体效应对烯丙基化反应的影响
确立了反应的最优溶剂后,随即探究了配体对该反应的影响(表2)。配体的轴手性和中心手性存在明显的匹配/错配现象,当使用(S)-BINOL衍生的配体L1时,产物的对应选择性仅有-37%ee,而当使用(R)-BINOL衍生的配体L2时,反应的对映选择性有明显的提高(-63%ee)。硫醚片段的电性和空间位阻同样对产物的对应选择性具有明显的影响。例如,含有吸电子基的对溴苯硫醚衍生配体L3使产物的对映选择性明显降低(-36%ee),具有相对较大空间位阻的2-甲基苯硫醚和2-甲氧基苯硫醚衍生的配体L4、L5能够使产物的对映选择性提高至-67%ee,而2,6-二甲基苯基硫醚衍生的配体L6则使产物的对映选择性大幅下降(-27%ee)。烷基硫醚结构的配体同样对反应的立体控制具有不利的影响,无论是使用叔丁基还是苄基(L7、L8)配体,反应的对映选择性均有所下降。苯酚与胺基硫醚合成的亚磷酰胺(L9)同样对反应具有一定的立体控制,但主产物的构型发生了变化(31%ee)。此外,商业可得的亚磷酰胺配体(L10)应用于该反应时,给出的反应效果均较差。

表2 配体结构对反应的影响
2.3 碱和温度对烯丙基化反应的影响
以L5作为最优配体,探究了碱和温度对反应的影响(表3)。使用碳酸钾作碱时,反应提高至-72%ee,但反应时间延长至8 h。使用BSA+KOAc、 BSA+LiOAc或三乙胺时,反应几乎不能发生。当反应在室温条件下时,产物的对映选择性提高至-76%,反应时间延长至24 h。反应温度继续降至0 ℃时,产物的ee值能够提高至85%,但是反应效率大幅度降低。

表3 对碱和反应温度筛选
以硫酚(醇)、2-氟苯甲醛和R-(α)-甲基苄胺为原料,首先合成了胺基硫醚,随后与手性联萘二酚通过模块组合快速得到了同时具有轴手性和中心手性的硫醚-亚磷酰胺配体,该类配体具有反应结构多样、易于修饰、水氧稳定和原料易得等特点,并对配体的核磁共振氢谱与碳谱、高分辨质谱、熔点和比旋光度进行了表征。对配体/钯络合物催化的不对称烯丙基化反应研究发现,当使用二氯甲烷作溶剂,碳酸铯作碱时,反应能够得出较好的结果。同时,该类配体存在轴手性和中心手性的匹配/错配现象,使用R-BINOL衍生的配体时,反应能够得到更好的结果。此外,硫醚片段的空间位阻和电子效应对配体也具有很大的影响,具有给电子芳基硫醚片段的配体能够使产物的立体选择性更好。在最优条件下,目标产物能够达到76%的对映选择性。目前,该类配体在其他金属催化的不对称合成中的应用仍在研究中。
