CRISPR基因组编辑技术在结核分枝杆菌中的研究进展及应用
2023-02-04曲芸墨丁鑫园闫玫漪郭晓鹏孙义成
曲芸墨 丁鑫园 闫玫漪 郭晓鹏 孙义成
近年来,感染耐药结核分枝杆菌甚至耐多药结核分枝杆菌的患者例数逐渐增加,这使得结核病的防治遭遇重大挑战。为了应对此挑战,有必要研发新型抗结核疫苗及抗结核药物,这需要系统揭示结核分枝杆菌的基因功能并阐明其致病机制。分枝杆菌尤其是结核分枝杆菌的遗传操作十分困难,传统的基因组编辑方法如利用穿梭质粒[1]、自杀质粒[2]和噬菌体转导[3]等工具费时费力且效率不高,极大地限制了对结核分枝杆菌的生物学研究。
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR associated protein)即成簇的规律间隔短回文重复序列和 CRISPR 相关蛋白,是细菌和古细菌抵抗病毒等外源遗传物质入侵的一种获得性免疫系统[4]。CRISPR 簇由众多短而保守的重复序列和间隔序列组成。Cas基因一般位于CRISPR序列两侧,是一类较大的多态性家族基因。在成熟的单一间隔序列 的crRNA(CRISPR RNA)形成之后, 其会与Cas蛋白组成一个复合物, 基因组上的原间隔序列(protospacer adjacent motif,PAM)可以帮助 crRNA/Cas 复合物在基因组上精准定位,而后通过 crRNA 上的间隔序列与靶标序列进行碱基配对,并引导Cas蛋白或蛋白复合物对基因片段进行剪切,造成DNA双链断裂(double strands break, DSB)[5]。
近年来,基于CRISPR-Cas开发的基因工程工具被广泛应用到真核生物和原核生物中,极大地推动了功能基因组学的研究。在分枝杆菌中,CRISPR-Cas相关的基因工程技术也得到了快速发展[6],包括CRISPR辅助的同源重组基因组编辑、CRISPR辅助的非同源末端连接基因敲除、单碱基编辑技术和CRISPRi等在内的几种方法的开发实现了高效的分枝杆菌基因组编辑和转录调控。笔者总结了近年来几种CRISPR-Cas辅助的基因组编辑技术在分枝杆菌中,特别是在结核分枝杆菌中的发展过程、应用及其优缺点(表1),并对新策略的发展进行了展望。
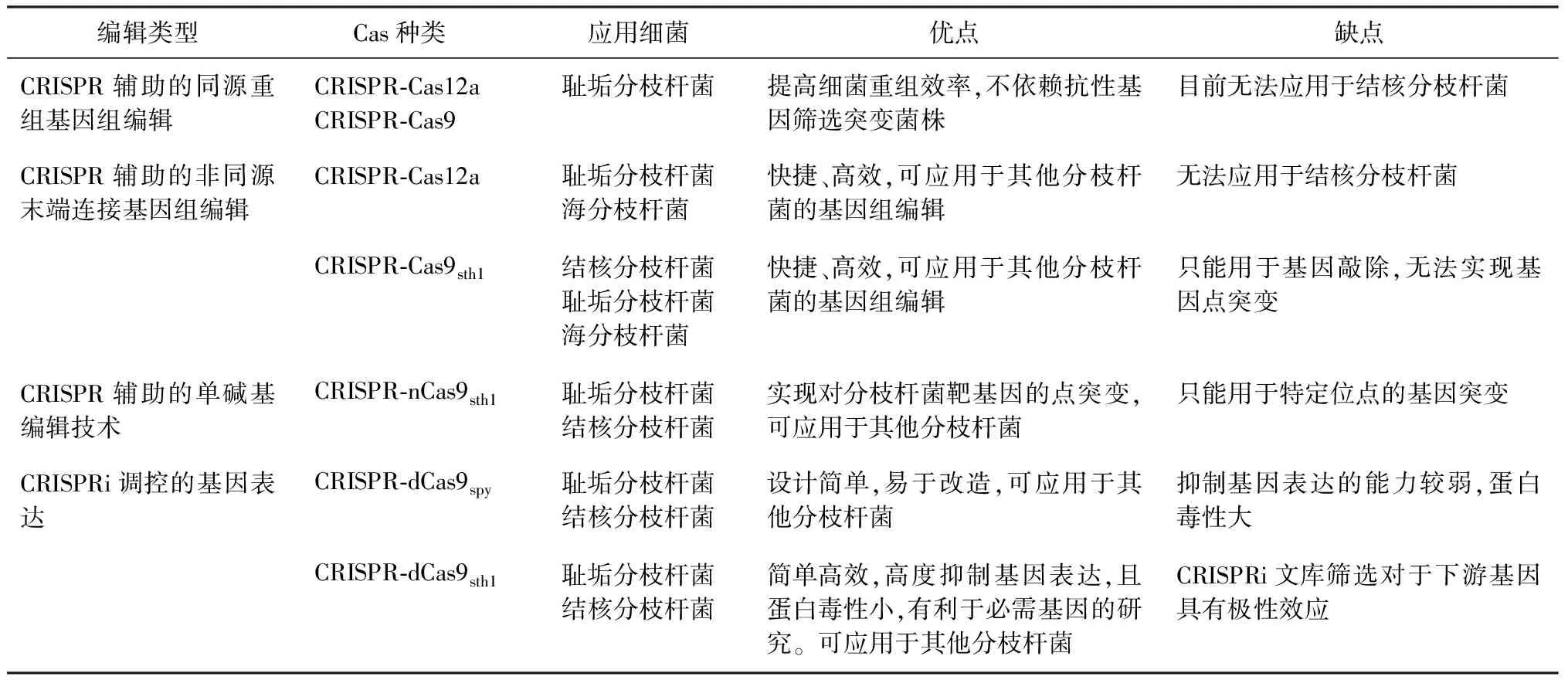
表1 结核分枝杆菌中的CRISPR基因组编辑技术
一、CRISPR-Cas辅助的同源重组基因组编辑技术
传统的同源重组(homologous recombination,HR)技术依赖细菌本身的 RecA蛋白介导的重组系统,将基因组序列与具有同源序列的外源DNA模板进行交换,实现定点整合。但是该方法通常需要很长的同源臂并且重组效率低。针对这一缺陷,van Kessel 等[7]在2007年开发了一种噬菌体重组蛋白辅助的重组策略,通过在细菌中高表达分枝杆菌噬菌体Che9c的gp60蛋白和gp61蛋白来提高重组效率。其中gp60具有5′~3′双链DNA外切酶活性,而gp61作为单链DNA退火蛋白,可以促进互补链 DNA 的退火、交换和重组。在乙酰胺诱导下同时表达这两个噬菌体蛋白,可以在耻垢分枝杆菌中实现高效率基因重组。虽然该方法效率很高,但必须使用抗生素抗性基因来筛选,而抗性基因的去除涉及多个实验步骤,包括利用位点特异性重组酶等,会使突变株构建的工作量增加。
CRISPR-Cas系统可以很好地解决这个问题,该系统利用人工合成的sgRNA将Cas蛋白引导到靶基因附近,并切割DNA造成双链断裂。由于Cas蛋白可以持续切割未发生编辑的细菌并使其死亡,直接起到了筛选作用,这样一来就可以避免抗生素抗性基因的使用,摒弃了去除抗性筛选基因的繁琐过程,因此,基于CRISPR和重组联用技术的开发可以成为细菌基因组编辑的优良工具,但是长期以来都没有成功应用于分枝杆菌。Yan等[8]于2017年利用来自弗朗西斯菌(Francisbacteria)的CRISPR-Cas12a系统,结合噬菌体蛋白gp60和gp61介导的同源重组系统,在耻垢分枝杆菌中实现了无标记的基因组高效编辑,包括基因插入、替换、删除及连续删除等常用基因操作。CRISPR-Cas12a是CRISPR系统中Ⅴ型效应蛋白,能够识别富含胸腺嘧啶核苷的 PAM 区(5′-YTN-3′),并且形成的 CRISPR-Cas12a 复合物不需要tracrRNA的参与。将Cas12a编码基因克隆在pJV53质粒上,由Pmyc1tetO启动子诱导表达,靶向目标序列的crRNA元件单独表达在一个温度敏感型质粒上,与带有同源臂的模板序列同时转入含有质粒pJV53-Cas12a并用乙酰胺提前诱导的耻垢分枝杆菌的感受态细胞中进行基因组编辑。当使用59-79nt的单链DNA作为修复模板时,可以高效的实现点突变,并可以有效进行 1000 bp 以下片段的删除;当使用带有500 bp双链DNA同源臂作为模板时,可以进行大片段的插入、删除及替换。此外,传统重组的方法需要每一步去除抗性筛选基因后再进行下一个基因的敲除,而CRISPR-Cas12a 辅助的重组系统由于辅助质粒很容易丢掉,通过交替使用含有不同抗性标签的 crRNA 质粒能够方便快捷地进行连续敲除,因此可以更方便地构建多个基因敲除的菌株。然而由于重组效率等方面的限制,目前该技术尚无法应用于包括结核分枝杆菌在内的其他分枝杆菌的基因组编辑。
二、CRISPR-Cas辅助的非同源末端连接基因组编辑技术
除了HR以外,非同源末端连接(non-homologous end joining, NHEJ)修复也是细胞内DSB的主要修复途径之一[9]。相比于同源重组,NHEJ更易于在DSB位点发生插入和(或)删除突变,并产生破坏目标基因的移码突变。但与真核生物的修复途径相比,大多数细菌缺乏内源 NHEJ 修复系统,主要通过 HR 途径对 DSB 修复[10]。目前发现少数细菌如分枝杆菌、铜绿假单胞菌和枯草芽孢杆菌等具有NHEJ修复通路。在2017年以前尚不能利用HR或NHEJ系统实现高效的结核分枝杆菌基因组编辑,因此当HR编辑暂时无法实现时,NHEJ系统的优化应用以实现高效基因敲除一直是研究人员重点关注的方向。Sun等[11]发现在耻垢分枝杆菌中利用Cas12a进行基因组剪切可以被 NHEJ系统修复,由此实现基因组编辑。但是该方法只能在耻垢分枝杆菌中发挥作用,而且效率较低。Yan等[12]发现Cas12a剪切引起的DSB在海分枝杆菌中也可以被NHEJ修复。进一步研究发现,海分枝杆菌中除kuC基因和ligD基因外,位于这两个基因之间的nrgA基因也可能参与了NHEJ修复。当将海分枝杆菌的NHEJ系统在耻垢分枝杆菌中异源高表达,联合CRISPR-Cas12a系统可以在耻垢分枝杆菌中发生基因组编辑,然而效率比较低。有研究显示,通过抑制RecA介导的同源重组途径,可以增加NHEJ修复的效率[13]。因此,通过在耻垢分枝杆菌中高表达RecA的抑制蛋白RecX或者其显性负性突变体RecAR60C抑制RecA介导的同源重组,同时高表达海分枝杆菌NHEJ系统,结合 CRISPR-Cas12a或者CRISPR-Cas9sth1(StreptococcusthermophilusCas9)系统可以在耻垢分枝杆菌实现高效的基因组编辑。进一步将该优化后的CRISPR-NHEJ系统应用到结核分枝杆菌中,通过高表达海分枝杆菌NHEJ系统、抑制RecA依赖的的同源重组修复途径,以及在稳定期产生DSB等方面的优化改进增强NHEJ的修复效率,利用CRISPR-Cas9sth1实现高效的基因组编辑。同时,进一步通过使用成对的sgRNA还在结核分枝杆菌中实现了大片段删除和双基因的同时敲除,可以在N+ 2个月内产生N个突变,而传统方法通常需要4N个月或更长时间,该方法为在这种缓慢生长的结核分枝杆菌中研究多个基因的功能提供了便利。
CRISPR-NHEJ技术在结核分枝杆菌中成功实现高效的基因组编辑,使得结核分枝杆菌突变文库的构建及应用成为可能。基于这项技术,我们利用该系统构建了一个针对结核分枝杆菌毒素-抗毒素系统的小型突变体库[12]。选择了44个毒素基因,共设计88个sgRNA,随后将其转到含有RecX辅助质粒和NHEJ相关元件的结核分枝杆菌感受态细胞中,最终一次转化可以得到3×104个转化子,其中大约85%的转化子发生基因组编辑。可见,可以利用此技术构建结核分枝杆菌的全基因组CRISPR突变文库,用于结核分枝杆菌的功能基因组学研究。
三、基于CRISPR系统的单碱基编辑技术
基于CRISPR-Cas系统辅助的重组的方法可以在耻垢分枝杆菌中实现基因的点突变[8],但是该系统尚无法在结核分枝杆菌中应用。近年来,另一种基于CRISPR的基因组编辑方法——单碱基编辑技术的开发为精确的基因操作提供了一种不需要引入DSB或供体DNA模板的新策略[14-15]。单碱基编辑系统主要包括胞嘧啶碱基编辑器和腺嘌呤碱基编辑器。胞嘧啶碱基编辑器由一个催化活性受损的Cas核酸酶、胞苷脱氨酶和尿嘧啶DNA糖基化酶抑制剂组成。核酸酶活性缺陷的Cas蛋白不能对DNA造成双链断裂,但能够将脱氨酶定位到靶标基因,脱氨酶随后在 PAM 位点附近的一个小编辑窗口区内将胞嘧啶转化为尿嘧啶,由此产生的错配双链DNA可以通过修复机制进行修复,从而实现胞嘧啶到胸腺嘧啶的精确单碱基替换[16]。此外,通过催化特殊密码子(CAA、CAG、CGA 或 TGG)的转换,CBE可用于通过生成提前终止密码子来破坏目标基因。
Ding等[17]将胞嘧啶碱基编辑技术应用于耻垢分枝杆菌和结核分枝杆菌中,可以实现对结核分枝杆菌的基因进行定点突变。首先构建一个碱基编辑器质粒pCBE,该质粒主要表达来自嗜热链球菌的核酸酶活性受损的nCas9sth1、胞苷脱氨酶APOBEC1和UGI形成的融合蛋白以及sgRNA作用元件。将pCBE单独转进耻垢分枝杆菌中进行基因组编辑时,仅有约10%的基因组编辑效率,并且在结核分枝杆菌中无法实现基因组编辑。作者推测细菌DNA修复通路,如同源重组和错配修复等可能干扰了单碱基编辑系统的作用。为此,通过高表达RecX抑制RecA介导的同源重组修复途径,在耻垢分枝杆菌中可以取得90%以上的单碱基编辑的效率。在结核分枝杆菌中,作者通过高表达RecX抑制同源重组修复途径,并进一步通过高表达错配修复蛋白NucS的显性负性突变体抑制错配修复途径,应用胞嘧啶碱基编辑系统,成功实现了在结核分枝杆菌基因组中进行胞嘧啶到胸腺嘧啶的碱基替换,效率为12.5%~75%。进一步利用该方法构建的katG突变株在异烟肼药物敏感性试验中表现出相比于野生型更高的耐药性,验证了单碱基编辑技术在探究细菌耐药方面的作用。
四、基于CRISPRi的基因表达调控
CRISPR-Cas辅助的同源重组和非同源末端连接技术可以对分枝杆菌实现高效的基因组编辑,但是这些方法不适合用于研究必需基因。CRISPR interference (CRISPRi)提供了一种简单且高效的基因表达调控策略。在CRISPRi中,Cas9 的2个核酸酶结构域RuvC和 HNH的突变(即dCas9)破坏了该酶的催化活性,但保留了其 RNA 引导的 DNA 靶向能力。dCas9-sgRNA复合物通过结合到目标基因上干扰其转录的延伸,从而导致序列特异性基因表达沉默[18]。
最开始用于分枝杆菌基因表达调控的CRISPRi系统使用的是来自化脓性链球菌的dCas9(dCas9spy)[19-21]。当将诱导表达dCas9spy的质粒和诱导表达靶向目标序列的sgRNA质粒转进耻垢分枝杆菌和结核分枝杆菌后,检测结果显示最高可以抑制目标基因表达的效率较低,而一些分枝杆菌基因需要高水平的抑制才能展现出相应表型,而且高表达Cas9spy对分枝杆菌生长有抑制作用。因此,为了开发更高效的CRISPRi系统,2017年Rock等[22]通过筛选来自不同种类细菌的CRISPR-dCas系统,并将密码子优化的dCas9-sgRNA和其他元件表达在一个质粒上,转进分枝杆菌中进行转录抑制实验,结果显示来自嗜热链球菌的CRISPR-dCas9(dCas9sth)展现了强大的基因沉默能力,通常可以实现内源性基因表达20~100倍的抑制,且蛋白毒性较小。这项发现实现了在耻垢分枝杆菌和结核分枝杆菌中的大范围应用[23-25]。McNeil等[23]利用该系统,研究了分枝杆菌叶酸生物合成途径基因的协同作用,并结合靶标两个相关基因的小分子药物,表明CRISPRi系统可以用于化学-遗传的相互作用研究。Adolph等[24]在2022年也利用CRISPRi分别抑制了sdhA1、sdhA2和frdA的一个、两个或三个基因的表达,发现sdhA1和sdhA2在琥珀酸氧化中功能冗余,只有同时抑制sdhA1和sdhA2的表达才能抑制结核分枝杆菌中的琥珀酸氧化。这些结果表明CRISPRi不仅可以方便研究结核分枝杆菌中的单个基因,而且可以用于研究多个基因的协同作用,为结核分枝杆菌生物学功能的研究提供了便利。
五、CRISPR-Cas系统在分枝杆菌高通量筛选中的应用
相比于其他高通量筛选技术,CRISPR文库筛选具有方便、可靠、易分析等多种优点,逐渐在生物学研究中得到广泛应用,而CRISPR文库筛选最近也应用于结核分枝杆菌研究。抑制必需基因的化合物,往往不能取得杀菌成效。其中一个重要原因为有些化合物只能部分抑制靶标基因功能,而某些被抑制的靶标基因其仅存的一点功能足以维持细菌生长。因此,好的药物靶标不仅是必需的基因,还应该是“脆弱的”基因,即被部分抑制就能造成细菌适应性明显下降。为了量化分枝杆菌基因的“脆弱性”, Bosch等[19]在2021年基于dCas9sth的全基因组CRISPRi筛选方法,利用不同PAM序列和不同sgRNA长度产生的抑制效率的差异来调控基因表达水平,分析了杀死细菌每个基因需要被抑制的程度。利用该方法,Bosch等量化了结核分枝杆菌和耻垢分枝杆菌中必需基因的脆弱性,并使用这些结果来确定分枝杆菌生理学中的限速步骤和鉴定潜在的药物靶点[19],这为抗结核药物的开发奠定了基础。此外,一些小型文库也被构建用于在各种条件下筛选与表型相关的基因。例如,McNeil等[23]在耻垢分枝杆菌中成功构建了同时靶向参与细菌呼吸的基因的两两组合的CRISPRi文库,用于确定呼吸复合物之间的合成致死和协同相互作用。与此同时,在感染模型中,Babunovic等[26]还利用CRISPRi技术筛选出了在全反式维A酸(ATRA)激活的巨噬细胞中,结核分枝杆菌生存所必需的基因。除了利用该系统进行筛选,McNeil和Cook[27]还证明了在快速验证药物靶点重要性上CRISPRi技术也有着很大优势。
目前,耐药结核病患者例数的逐渐增加引起科学家的重点关注,为了找到更多的药物及药物靶点,Li等[28]在2022年将研究目标锁定在利用CRISPRi技术探究影响已有抗结核药物的化学-基因相互作用。研究人员构建了几乎覆盖所有结核分枝杆菌基因的CRISPRi文库,并用9种药物(包括7种抗结核临床代表药物和2种非结核药物)通过化学-遗传相互作用进行筛选,测序分析sgRNA丰度进而确定相关基因。首先发现利福平、万古霉素和贝达喹啉的常见致敏靶点是结核分枝杆菌细胞膜的主要成分分枝菌酸-阿拉伯半乳糖苷-肽聚糖(mAGP)复合物,mAGP可能会通过改变胞膜通透性来影响药物杀菌作用。随后筛选了靶向核糖体的药物耐药的相关性位点,结果提示核糖体甲基转移酶TsnR很可能与利奈唑胺耐药有关。该研究还发现ettA的突变会导致结核分枝杆菌对多种抗结核药物低水平耐药[28],这可能是南美等多个国家多耐药结核病暴发的原因之一。此外,还发现氨基糖苷类等多种药物未知的潜在耐药机制,甚至首次发现了一类针对大环内酯类药物获得性敏感的结核分枝杆菌亚系,提出可以采用克拉霉素对这些菌株进行抗结核治疗。采取同样的策略,Poulton等[29]也利用了CRISPRi筛选技术分析了PBTZ169的化学-遗传相互作用。PBTZ169是一种具有较大应用前景的苯并噻嗪酮类抗结核药物,作用靶点是参与结核分枝杆菌阿拉伯半乳聚糖合成的酶亚基DprE1。筛选结果发现,负向调控编码外排泵基因mmpS5-mmpL5的rv0678基因的抑制导致PBTZ169耐药。Poulton等[29]进一步利用rv0678突变株和mmpS5-mmpL5突变株进行耐药机制的验证,最终证明rv0678突变的确与PBTZ169和另一种苯并噻嗪酮类抗结核药物BTZ043耐药相关。这一结果提示,在PBTZ169和BTZ043临床试验中监测临床rv0678突变菌株的感染率的重要性,也进一步体现了利用CRISPRi系统高通量筛选的有效性及可靠性。
虽然CRISPR 敲除 (CRISPR-knockout, CRISPR-KO)在真核生物的高通量筛选中已经得到广泛应用,然而由于在细菌等原核生物中敲除效率较低,在细菌中尚未得到大范围的应用。Yan等[30]在前期研究中建立的CRISPR-NHEJ基因组编辑具有操作简单、高效的特点,在2022年利用该技术成功构建了结核分枝杆菌全基因组范围内的CRISPR-KO文库。该文同时参考Rock等[22]的研究构建了结核分枝杆菌的CRISPRi文库,将这两个文库组合应用,首先分析了结核分枝杆菌的必需基因。研究结果证实了CRISPR-KO文库筛选的可靠性,同时发现CRISPR-KO和CRISPRi文库筛选各自具有独特的优势和不足。CRISPRi文库筛选对于下游基因具有极性效应,如果在某个必需基因的下游存在非必需基因,那么该非必需基因很有可能会被误认为是必需基因,而CRISPR-KO在筛选中则不会出现这个现象。利用CRISPRi抑制基因表达时具有靶向链的选择性,只有当靶向非模板链时才能表现出较好的抑制作用,而CRISPR-KO没有这方面的限制。文库的实际构建中一般不能实现百分之百的全覆盖,当CRISPR文库无法完全覆盖时,CRISPRi可能会将缺失的必需基因识别为非必需基因,而CRISPR-KO可能将非必需基因识别为必需基因。所以,这些性质表明两者在筛选方面有着各自的优势和不足,将他们联合应用将得到更全面、可靠的筛选结果。该研究进一步利用这两个文库进行了抗结核药物贝达喹啉的化学-遗传相互作用的研究(图1)。首先构建了覆盖全基因组的CRISPRi和CRISPR-KO的结核分枝杆菌文库,抗结核药物贝达喹啉处理后,通过二代测序分析文库中sgRNA的丰度,分析确定影响贝达喹啉杀菌能力的基因,具体见图1。测序结果发现数十个与贝达喹啉耐药性或者是易感性相关的基因,包括已知的编码外排泵的基因mmpS5-mmpL5和贝达喹啉靶标的ATP合成相关通路的基因。整体分析结果表明,CRISPRi文库在筛选能影响贝达喹啉杀菌的必需基因时更有优势,而CRISPR-KO文库在筛选能影响贝达喹啉杀菌的非必需基因时更加灵敏。挑选具有小分子抑制剂的2个基因topA和lprG,通过突变株构建,验证了2个筛选得到的基因topA、lprG影响贝达喹啉杀菌能力;通过药物协同杀菌实验等,发现这2个基因的靶标小分子AMSA、LB04-Ⅲ确实与贝达喹啉具有协同杀菌作用。根据研究结果初步揭示了这些影响贝达喹啉杀菌的相关基因作用机制。这项研究说明组合CRISPR文库筛选,能够互补各自的缺点,整合两种筛选系统的优势,能更加全面地了解结核分枝杆菌内部的生物学过程,并在未来应用于更多细菌中的高通量研究。
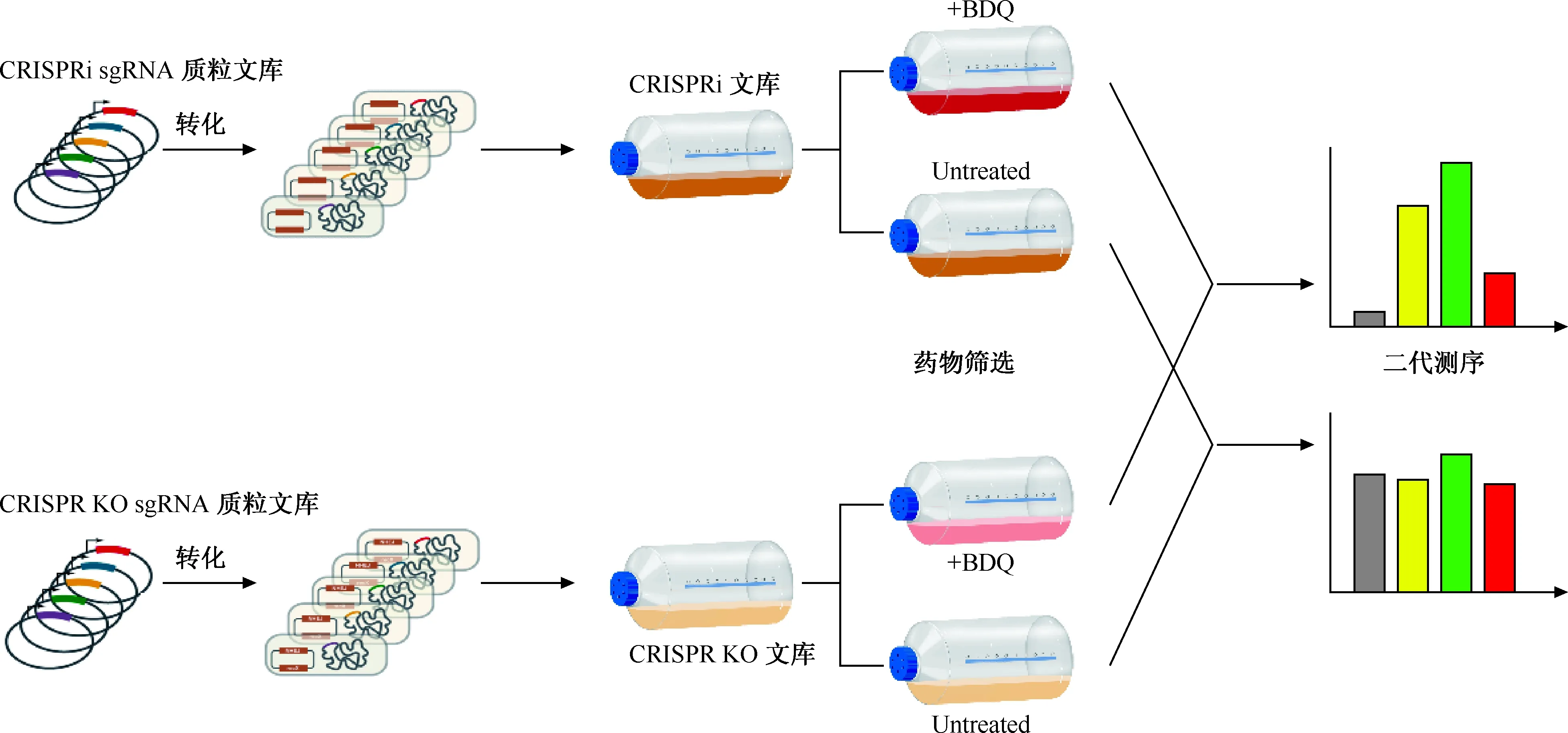
图1 利用CRISPRi与CRISPR-KO组合文库研究化学-基因相互作用研究
六、总结与展望
如今结核病对人类社会还存在着很大威胁,迫切需要进一步加深对结核分枝杆菌生理、毒力和耐药等相关基因的研究和认识,开发新的抗结核药物或疫苗。CRISPRi技术凭借着简单、快速和低成本等优点展现出很大应用潜力和优势,使得必需基因的研究成为可能。CRISPR-NHEJ介导的基因组编辑技术使分枝杆菌突变体的构建更加简单、快捷,这进一步方便了分枝杆菌相关基因的生物学功能研究。基于CRISPR的CRISPR-KO和CRISPRi突变文库的高通量筛选的应用将极大促进结核分枝杆菌功能基因组学的相关研究。
目前,CRISPR辅助的同源重组技术虽然可用于耻垢分枝杆菌基因组的基因组编辑,但是该系统尚无法成功应用到结核分枝杆菌中,这在未来值得进一步改进和探究。此外,虽然目前单碱基编辑器在耻垢分枝杆菌中实现高效率的基因组编辑,但是其在结核分枝杆菌中的编辑效率较低,这项技术还有待于进一步的优化和改造。
综上,基于CRISPR的结核分枝杆菌基因组编辑技术的开发及优化研究,将极大促进结核分枝杆菌的基础研究,加深对结核分枝杆菌基本生物学功能的认识,有助于抗结核药物及疫苗的研发,由此为结核病的防治策略提供理论依据和重要技术支持。
利益冲突所有作者均声明不存在利益冲突
作者贡献曲芸墨和丁鑫园:查阅文献、整理资料和撰写文稿;闫玫漪和郭晓鹏:文稿修改和编辑;孙义成:文章概念提出、资金支持、文稿修订和审核
