Analyses and identifications of quantitative trait loci and candidate genes controlling mesocotyl elongation in rice
2023-02-03ZHANGXijuanLAlYongcaiMENGYingTANGAoDONGWenjunLlUYouhongLIUKaiWANGLizhiYANGXianliWANGWenlongDlNGGuohuaJlANGHuiRENYangJlANGShukun
ZHANG Xi-juan ,LAl Yong-cai ,MENG Ying ,TANG Ao,DONG Wen-jun,LlU You-hong,LIU Kai,WANG Li-zhi,YANG Xian-li,WANG Wen-longDlNG Guo-hua,JlANG Hui,REN Yang,JlANG Shu-kun,
1 Institute of Crop Cultivation and Tillage,Heilongjiang Academy of Agricultural Sciences,Heilongjiang Provincial Key Laboratory of Crop Physiology and Ecology in Cold Region,Heilongjiang Provincial Engineering Technology Research Center of Crop Cold Damage,Harbin 150086,P.R.China
2 Northeast Branch of National Salt-Alkali Tolerant Rice Technology Innovation Center,Harbin 150086,P.R.China
3 Qiqihar Branch,Heilongjiang Academy of Agricultural Sciences,Qiqihar 161006,P.R.China
Abstract Rice direct seeding has the significant potential to save labor and water,conserve environmental resources,and reduce greenhouse gas emissions tremendously.Therefore,rice direct seeding is becoming the major cultivation technology applied to rice production in many countries.Identifying and utilizing genes controlling mesocotyl elongation is an effective approach to accelerate breeding procedures and meet the requirements for direct-seeded rice (DSR) production.This study used a permanent mapping population with 144 recombinant inbred lines (RILs) and 2 828 bin-markers to detect quantitative trait loci (QTLs) associated with mesocotyl length in 2019 and 2020.The mesocotyl lengths of the rice RILs and their parents,Lijiangxintuanheigu (LTH) and Shennong 265 (SN265),were measured in a growth chamber at 30°C in a dark environment.A total of 16 QTLs for mesocotyl length were identified on chromosomes 1(2),2(4),3(2),4,5,6,7,9,11(2),and 12.Seven of these QTLs,including qML1a,qML1b,qML2d,qML3a,qML3b,qML5,and qML11b,were reproducibly detected in both years via the interval mapping method.The major QTL,qML3a,was reidentified in two years via the composite interval mapping method.A total of 10 to 413 annotated genes for each QTL were identified in their smallest genetic intervals of 37.69 kb to 2.78 Mb,respectively.Thirteen predicted genes within a relatively small genetic interval (88.18 kb) of the major mesocotyl elongation QTL,qML3a,were more thoroughly analyzed.Finally,the coding DNA sequence variations among SN265,LTH,and Nipponbare indicated that the LOC_Os03g50550 gene was the strongest candidate gene for the qML3a QTL controlling the mesocotyl elongation.This LOC_Os03g50550 gene encodes a mitogen-activated protein kinase.Relative gene expression analysis using qRT-RCR further revealed that the expression levels of the LOC_Os03g50550 gene in the mesocotyl of LTH were significantly lower than in the mesocotyl of SN265.In conclusion,these results further strengthen our knowledge about rice’s genetic mechanisms of mesocotyl elongation.This investigation’s discoveries will help to accelerate breeding programs for new DSR variety development.
Keywords: japonica rice,direct-seeded rice (DSR),mesocotyl elongation,quantitative trait loci,candidate gene
1.lntroduction
Rice (Oryza sativaL.) is a staple food and nutritional source worldwide and fulfills the food security needs of more than half of the global population.Transplanted rice (TPR) systems are the major planting technique for rice production in Asia,and nearly 85% of rice is grown under such conditions in China (Liuet al.2015).Since climate change,urbanization,water shortages,and labor scarcity have become increasingly severe in many places,conventional transplanted rice production is facing increasing challenges (Sagareet al.2020).Compared to TPR systems,rice direct seeding is emerging as an efficient,labor-and water-saving,resource-conserving,mechanized,climate-smart,and economically viable cultivation technology in many countries (Farooqet al.2011;Liuet al.2015;Sagareet al.2020;Wanget al.2022).Although direct-seeded rice (DSR) has so many benefits over TPR,a low seedling emergence rate,poor seedling establishment,weed infestation,and high crop lodging are still the major limiting factors that obstruct its adoption (Farooqet al.2011;Liuet al.2015).Among these limiting factors,the difficulty of early uniform emergence and seedling establishment are critical determinants related to high yields in DSR.The mesocotyl is a crucial embryonic organ between the coleoptile node and the base of the radicle root,and it plays a critical role in rice seedling establishment.Its elongation during germination pushes the shoot tip to emerge out of the soil (Zhanet al.2020).Previous studies indicated that a long mesocotyl is essential for the early uniform emergence and early vigor of DSR (Sagareet al.2020).Mesocotyl elongation has been recognized as one of the most important traits for the breeding of DSR (Huanget al.2010;Lvet al.2020a).
Many studies have revealed that mesocotyl elongation is controlled by environmental factors and phytohormones(Choiet al.2003;Kumar and Ladha 2011).As the major environmental factor affecting plant development,light is a key factor controlling mesocotyl elongation(Chenet al.2004).Generally,mesocotyl elongation is enhanced in darkness and inhibited after light exposure(Lvet al.2020b;Zhanet al.2020).Temperature is another factor regulating mesocotyl elongation.The mesocotyl elongation ability differed according to different temperatures (15 to 45°C),and 30°C was reported as the optimal temperature in sorghum (Radford and Henzell 1990).Moreover,several plant hormones are also reported to influence mesocotyl elongation.The applications of exogenous gibberellins (GA),brassinosteroid (BR),ethylene (ETH),and cytokinin (CTK)are beneficial to cell elongation in mesocotyl (Takahashi 1973;Nguyenet al.2000;Caoet al.2005;Huanget al.2010;Lianget al.2016;Xionget al.2017;Sunet al.2018).In contrast,abscisic acid (ABA),jasmonic acid (JA),and strigolactones (SLs) inhibit mesocotyl elongation (Watanabeet al.2001;Huet al.2010;Huet al.2014;Xionget al.2017;Sunet al.2018).All of these phytohormones,except ETH,directly influence mesocotyl elongation by affecting either cell division or cell elongation.ETH works as a signal to regulate cell elongation through the JA biosynthesis pathway (Zhanet al.2020).
Dissection of the genetic basis of mesocotyl elongation is essential for a better understanding of the early seedling establishment ability of DSR.Recent studies have further revealed that mesocotyl elongation is controlled by quantitative trait loci (QTLs) in rice (Cai and Morishima 2002;Caoet al.2002;Ouyanget al.2005;Huanget al.2010;Leeet al.2012a,b).A previous study has identified five QTLs on chromosomes 1,2,3,6,and 11 using an F6population from Lijiangxintuanheigu (LTH)/Shennong 265 (SN265) under water and GA germination conditions (Huanget al.2010).A total of five QTLs for mesocotyl length were detected on chromosomes 1,3,7,9,and 12 by using a backcross inbred line(BIL) population from a cross between Kasalath and Nipponbare in two independent experiments (Leeet al.2012b).Two QTLs,qML1andqML3,were mapped on chromosomes 1 and 3 in an F8population from a cross between Ilpumbyeo and PBR (photoblastic rice) (weedy rice) (Leeet al.2012a).Eight QTLs for rice mesocotyl length were identified on chromosomes 1,3,6,7,8,and 12 using the doubled haploid (DH) population from IR64/Azucena (Caoet al.2002).Eleven QTLs were identified on chromosomes 1,3,4,5,6,9,and 11 in a recombinant inbred line (RIL) derived from a cross between Pei-kuh(indica) and W1944 (O.rufipogon) (Cai and Morishima 2002).Six QTLs with significant additive effects were detected on chromosomes 1,5,and 9 in a RILs population from Zhenshan 97B/Miyang 46 (Ouyanget al.2005).
As the cost of genome sequencing decreases,genome-wide association studies (GWAS) based on single-nucleotide polymorphisms (SNPs) have been widely adopted to find significant loci associated with mesocotyl elongation in rice (Wuet al.2015;Luet al.2016;Sunet al.2018;Zhaoet al.2018;Liuet al.2020).Three significantly associated loci for mesocotyl length were identified among a diverse worldwide collection of 510O.sativaaccessions using the GWAS method (Sunet al.2018).Seventeen major loci explaining~19.31%of the observed phenotypic variation were identified on chromosomes 1,4,5,6,7,9,and 11 based on 4 136 SNPs from a set of 469 globally diverseindicaaccessions(Luet al.2016).Thirteen loci controlling mesocotyl length were detected on rice chromosomes 1,3,4,5,6,and 9 using a set of 270 rice accessions (Wuet al.2015).Sixteen unique loci were identified on chromosomes 1,2,3,4,5,6,7,8,9,and 12,based on a mixed linear model(MLM) in a population of 208 rice accession (Liuet al.2020).Thirteen QTLs for mesocotyl length were detected in a set of 621 cultivated rice accessions from the 3 000 Rice Genomes Project (3KRGP) (Zhaoet al.2018).
Although so many QTLs controlling mesocotyl elongation have been detected,only a few were finemapped and cloned.GY1was the first gene controlling mesocotyl elongation clonedviamap-based cloning technology,and it encodes a PLA1-type phospholipase and functions at the initial step of JA biosynthesis to repress mesocotyl elongation in rice (Xionget al.2017).OsGSK2,a GSK3-like kinase involved in brassinosteroid signaling,was identified to control rice mesocotyl length by using the GWAS method.Molecular analyses showed thatOsGSK2regulates mesocotyl length by coordinating strigolactone and brassinosteroid signaling (Sunet al.2018).Knocking outOsPAO5 viaCRISPR/Cas9 technology can remarkably increase mesocotyl length in the rice cultivar Nipponbare.Further results revealed thatOsPAO5promotes mesocotyl elongation by synthesizing more ethylene and releasing less H2O2(Lvet al.2020b).The QTLqML1controlling mesocotyl length was identified on chromosome 1 as a wild genotype ofsd1(green revolution gene) as in Kasalath (Lv 2020).TheOsBAK1gene encoding a BRI1-associated receptor kinase (BAK1)also participates in regulating mesocotyl elongation (Yuanet al.2017).
The above information informs the need to identify more major and key mesocotyl elongation genes to develop new rice varieties better suiting DSR breeding practices,although multiple essential forward advances of rice mesocotyl elongation investigations have been achieved.The objectives of this study were (1)dissecting the genetic basis of mesocotyl elongation,(2) fine-mapping the quantitative trait loci controlling mesocotyl elongation ability,and (3) investigating the candidate genes of major QTLs for further research on the regulatory mechanisms of mesocotyl elongation.The discoveries from this investigation will provide rice breeders with more efficient breeding tools to accelerate breeding programs for new DSR variety development.
2.Materials and methods
2.1.Mapping population and its high-density linkage map
SN265 and LTH,twojaponicarice varieties with significant differences in mesocotyl elongation characteristics,were utilized to produce mapping populations.The female parental variety LTH is a traditional,tall,low-yielding landrace with a very short mesocotyl,whereas SN265 is a super-high-yielding cultivar with a long mesocotyl.The F11-mapping population consisting of 144 recombinant inbred lines (RILs) was developed from the cross between LTH and SN265viathe single-seed descent method (Jianget al.2020a).
We built a high-density bin map using the RIL population with a resequencing approach.The effective sequencing depths of the LTH and SN265 genomes were approximately 19-and 17-fold coverage,respectively.The overall effective depth coverage of these RIL lines ranged from 5.98-to 9.73-fold.A total of 2 818 bin markers were assigned to 12 rice chromosomes,in which each chromosome contained 186 to 313 bin markers.The total genetic distance of this linkage map was 2 840.12 cM,with an average genetic distance of 1.01 cM.The corresponding physical intervals of the adjacent bin markers were between 15.00 kb and 3.60 Mb,with a mean value of 128.80 kb (Jianget al.2020b).The raw sequence data of the two parents and each RIL have been deposited in the NCBI Short Read Archive with accession number PRJNA587802.
2.2.Evaluation of mesocotyl elongation ability in two years
The parents and their RIL population were grown on the agricultural farm of the Institute of Crop Cultivation and Tillage,Heilongjiang Academy of Agricultural Sciences.Field management practices were performed according to the most commonly followed agricultural practices of local farmers.The high-quality grains were harvested and retained to dry for three months and then stored at 5°C to maintain a relative humidity of approximately 10% (Jianget al.2020b).
We used the Petri dish method to evaluate the variation in mesocotyl elongation according to the descriptions by Huanget al.(2010).The fully-filled and healthy seeds were sterilized with 0.1% mercury chloride solution for approximately 10 min and rinsed 3 to 4 times with deionized water.Thirty seeds of each line were placed on two layers of filter paper in a Petri dish (90 mm diameter×10 mm height),and the dish was filled with 8 mL of distilled water.The Petri dish was wrapped in aluminum foil and transferred into a growth chamber at 30°C in a dark environment.The mesocotyl length was measured two times (2019 and 2020) independently.Three replications were set up every year.On the 10th day after the start of incubation,the mesocotyl length of each seedling was measured with a metric ruler (Huanget al.2010;Leeet al.2012b).We used the soil covering method to evaluate the mesocotyl elongation ability of the twoqML3a-NILs.The seeds of SN265,LTH,and two NILs were buried at a depth of 2.5 cm soil in a plastic box(length 20 cm,width 15 cm,height 8 cm).The plastic box was kept in a plant growth chamber with alternating temperatures of 30 and 26°C (14 h/10 h).Five days after sowing,the seedlings were carefully excavated and washed.The mesocotyl length was measured according to the distance from the base of the radicle root to the coleoptile node (Leeet al.2012b).
2.3.QTL mapping for mesocotyl elongation ability
To identify the QTLs for mesocotyl elongation in the RILs,we used the Haley-Knott regression method (HK)and the composite interval mapping (CIM) method in the R/qtl package (Bromanet al.2003).The threshold levels of HK and CIM were ensured by 1 000 permutations,and the QTL confidence interval was estimated using a 1.5-LOD drop from the peak LOD.The mean value of three replicates was used to measure each line to detect the QTLs.The percentage of the total phenotypic variance accounting for each QTL in the HK regression method was estimated by 1-10-2LOD/n,wherenis the sample size and LOD is the LOD score (Broman and Sen 2009).The percentage of the total phenotypic variance accounting for each QTL in the composite interval mapping method was estimated by thefitqtlfunction (Bromanet al.2003).We also compared the QTLs for mesocotyl elongation in this study to those detected in previous studies in accordance with the physical location in MSU 7.0 rice genome.
2.4.Annotation of genes for each quantitative trait locus interval
The annotation of genes for each QTL interval was performed using the MSU Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu).All SNPs and InDels of DNA sequences between SN265 and LTH were detected using the re-sequenced genome data described by a previous report (Jianget al.2017).The whole-genome sequence of each gene in the QTL region from SN265 (long mesocotyl) was further compared with Nipponbare (short mesocotyl) to ensure the high confidence of candidate genes for the QTLqML3a.A 364.45-Mb high-quality genome of SN265 with a contig N50 value of 6.96 Mb was provided by Liet al.(2018).The genome sequence of Nipponbare (version_7.0) was downloaded from the MSU Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu/pub/data/) (Kawaharaet al.2013).
2.5.Development of the NlLs for qML3a and the qRT-PCR analysis of LOC_Os03g50550
An F6plant,heterozygous at theqML3aregion on chromosome 3 but homozygous in the other regions,was selected based on the genotyping results of 114 SSR markers as described by a previous report (Jianget al.2012).Consequently,two NILs ofqML3awere selected from the descendant lines of the F6plant according to the re-sequence genotype (Jianget al.2020b).Two parents(SN265 and LTH) and twoqML3a-NILs (NIL-qML3aand NIL-qml3a) were used to identify the expression levels ofLOC_Os03g50550.The total RNA was extracted using a TRIzol reagent.The qRT-PCR analysis was executed using the SYBR FAST qPCR (KAPA) Kit.The qPCR primer pairs sequences forLOC_Os03g50550were 5´-TTCAACAGAGAGTAATGGCCG-3´ (forward) and 5´-TACCACAGCCGTCTCTCCAT-3´ (reverse).
3.Results
3.1.Phenotypic variation of mesocotyl elongation
SN265 has a much longer mesocotyl than LTH (Fig.1-A),as described in a previous report (Huanget al.2010).The mesocotyl length (mean±SD) of SN265 was (17.64±6.91) mm in 2019 and (13.13±6.20) mm in 2020,whereas the mesocotyl length of ‘LTH’ was (4.91±2.39) mm in 2019 and (1.79±0.86) mm in 2020 (Fig.1-B;Table 1).In 2019,the mesocotyl length variation of these RILs ranged from 2.72 to 28.08 mm,with an average value of (10.54±5.44) mm(Fig.1-D;Table 1).In 2020,the distribution of the mesocotyl length ranged from 0.64 to 18.69 mm (Fig.1-E;Table 1).Repeatable mesocotyl length results were obtained from both years for the investigation with a significantly positive correlation coefficient (r=0.52,P<0.001) (Fig.1-C).The mesocotyl length was also nearly normally distributed in the two years.These results strongly suggest that mesocotyl elongation is mainly controlled by genetic factors,and the variations between years are due to influences from environmental conditions.
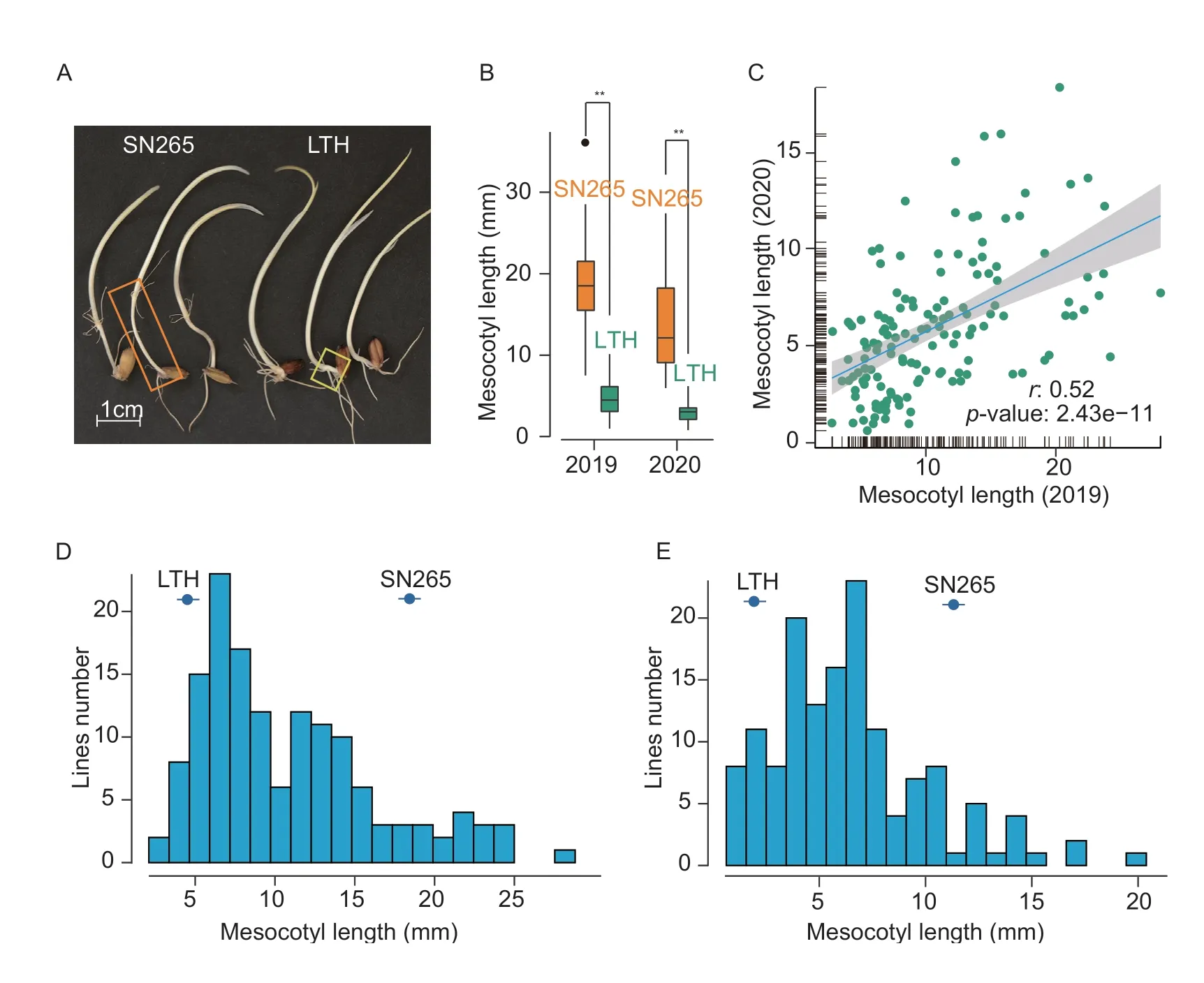
Fig.1 The mesocotyl elongation ability of the two parents and their RILs in 2019 and 2020.A,the mesocotyl length of Shennong 265(SN265) and Lijiangxintuanheigu (LTH).B,the mesocotyl lengths of SN265 (yellow) and LTH (green) on the 10th day under the temperatures of 30°C in 2019 and 2020.**,P<0.01,using Student’s t-test.C,the positive correlation of mesocotyl length investigated in 2019 and 2020.D,the mesocotyl length frequency distribution of the RILs in 2019.E,the mesocotyl length frequency distribution of the RILs in 2020.The strikethrough dots in (D) and (E) indicate the average mesocotyl length of LTH or SN265.
3.2.QTLs controlling mesocotyl length identified in 2019
Interval mapping (IM) results: A total of 13 QTLs for mesocotyl elongation were identified on chromosomes 1(2),2(2),3(2),4,5,7,9,11(2),and 12viathe interval mapping method (HK method) (Fig.2-A;Table 2).The QTL confidence intervals covered the genomic physical distance from 0.09 to 2.78 Mb with the corresponding genetic distance from 0.40 to 8.58 cM.The additive effects of these QTLs on mesocotyl elongation ranged from 1.76 to 3.57 mm.The explained phenotypic variation of these QTLs varied from 10.50 to 43.44%.QTLsqML1a,qML3a,qML3b,qML5,qML7,qML9,qML11a,andqML12had a positive allele from SN265,whereas the other five QTLs (qML1b,qML2a,qML2d,qML4,andqML11b) had a positive allele from LTH.Four major QTLs,qML1a,qML1b,qML3a,andqML3b,had more than 20% phenotypic variation (Table 2).
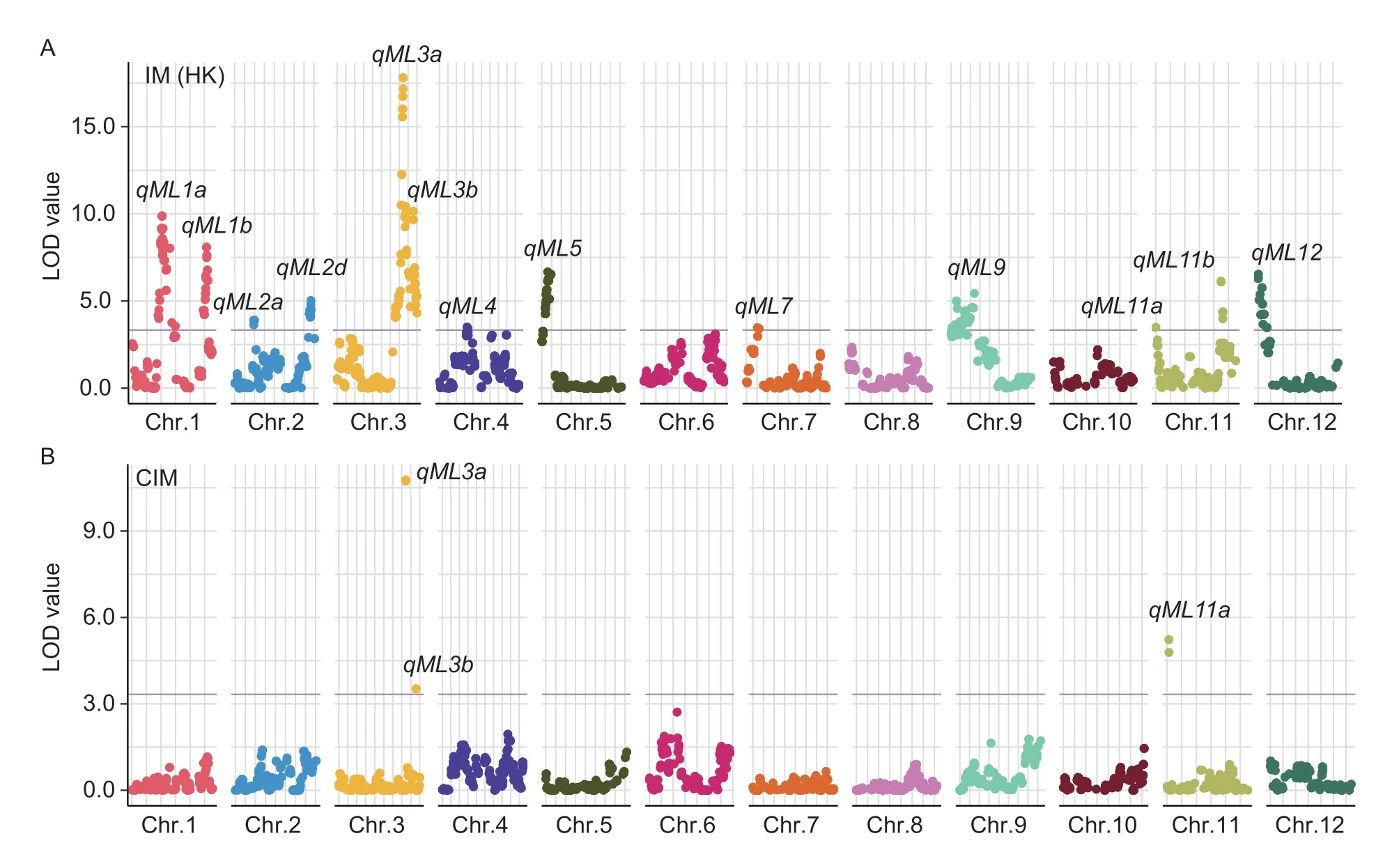
Fig.2 The genomic locations of the QTLs controlling mesocotyl elongation ability in the RILs in 2019.A,13 QTLs for mesocotyl elongation ability were identified in the RIL population via the interval mapping method.B,three QTLs for mesocotyl elongation ability were identified in the RIL population via the composite interval mapping method.

Table 1 Performance of mesocotyl elongation in Shennong 265 (SN265),Lijiangxintuanheigu (LTH),and their RIL population
Composite interval mapping (CIM) results: Only three QTLs (qML3a,qML3b,andqML11a) were reidentified in this RIL population by using the composite interval mapping method (Fig.2-B;Table 3).The QTLqML3awas redetected with the largest LOD value of 10.78 and explained 18.23% of the phenotypic variation,whileqML3bandqML11awere found to be minor QTLs with LOD values of 3.96 and 5.54,corresponding to 5.99 and 8.58% of the explained phenotypic variations,respectively.The confidence intervals ofqML3bandqML11ashrank from 0.09 and 1.24 Mb to 0.09 and 0.32 Mb,respectively.The additive effects ofqML3a,qML3b,andqML11aon mesocotyl elongation ranged from 1.59 to 2.71 mm.None of the QTLs with a positive allele from LTH were reidentified.
3.3.QTLs controlling mesocotyl length identified in 2020
IM results: Nine QTLs controlling mesocotyl elongation were identified on chromosomes 1(2),2(2),3(2),5,6,and 11 (Fig.3-A;Table 2).The QTL confidence intervals covered the genomic physical distance from 0.10 to 3.47 Mb with the corresponding genetic distance from 0.40 to 12.02 cM.The additive effects of these QTLs on mesocotyl elongation ranged from 1.14 to 1.66 mm.The explained phenotypic variations of these QTLs varied from 10.07 to 22.40%.QTLsqML1a,qML3a,qML3b,andqML5had a positive allele from SN265,whereas the other five QTLs (qML1b,qML2c,qML2d,qML6,andqML11b)had a positive allele from LTH.Only theqML3aQTL was responsible for more than 20% of the phenotypic variation and was identified as a major QTL (Table 2).
CIM results: Two QTLs,qML2candqML3a,were reidentifiedviathe composite interval mapping method(Fig.3-B;Table 3).The major QTLqML3awas redetected with the largest LOD value of 9.85 and explained 21.94%of the phenotypic variation,whileqML2cwas identified as a minor QTL with LOD values of 6.51,corresponding to 13.71% phenotypic variation.A novel QTL,qML2b,was discovered on chromosome 2 with a LOD value of 4.70 and 9.59% of the explained phenotypic variation.The confidence interval of theqML2cQTL shrank from 0.91 to 0.50 Mb.The additive effects ofqML3a,qML2b,andqML2con mesocotyl elongation ranged from 1.31 to 1.75 mm.The novel QTLqML2bhas a positive allele from SN265.
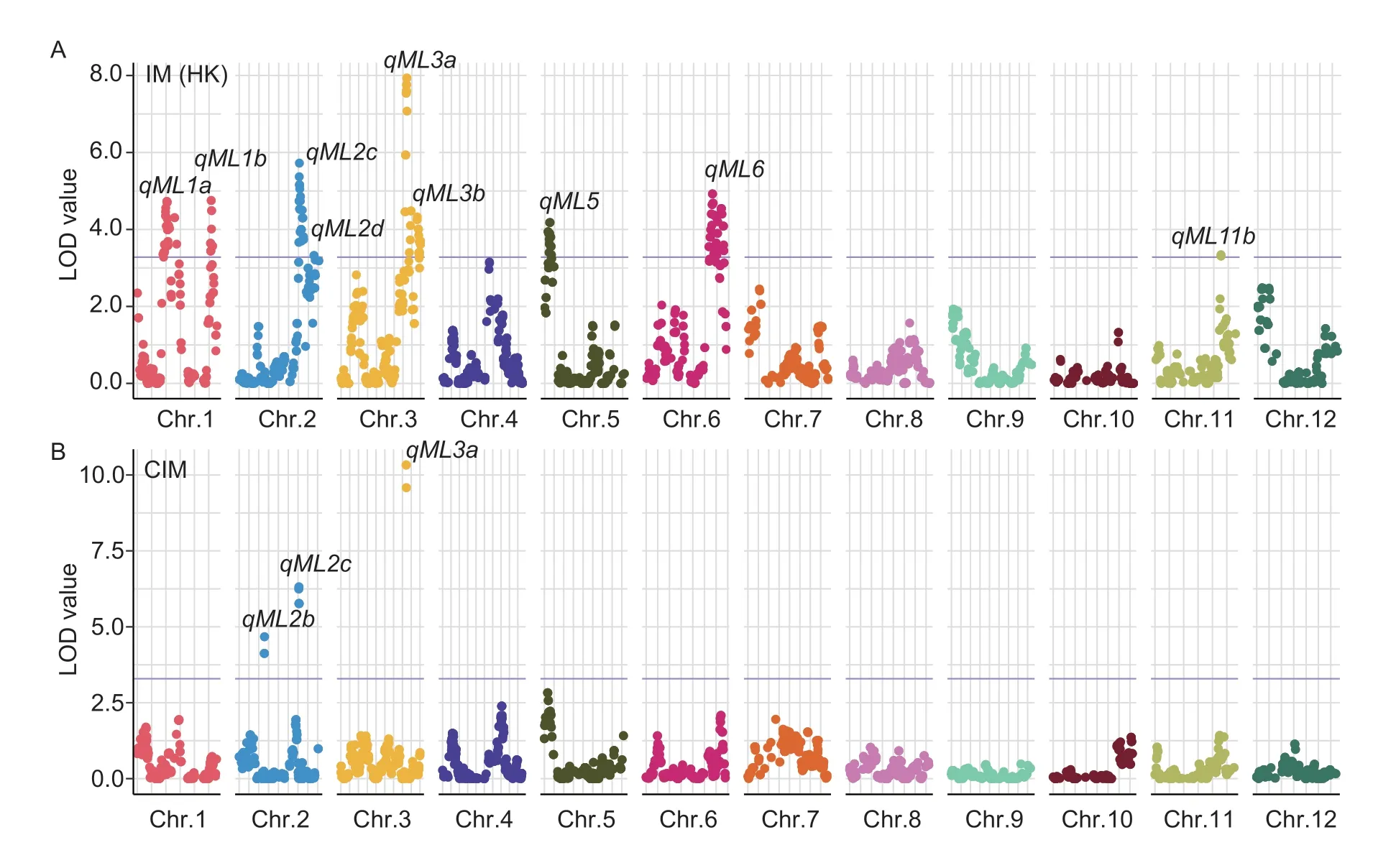
Fig.3 The genomic locations of the QTLs controlling mesocotyl elongation ability in the recombinant inbred lines (RILs) in 2020.A,eight QTLs for mesocotyl elongation ability were identified in the RIL population via the interval mapping method.B,three QTLs for mesocotyl elongation ability were identified in the RIL population via composite interval mapping method.
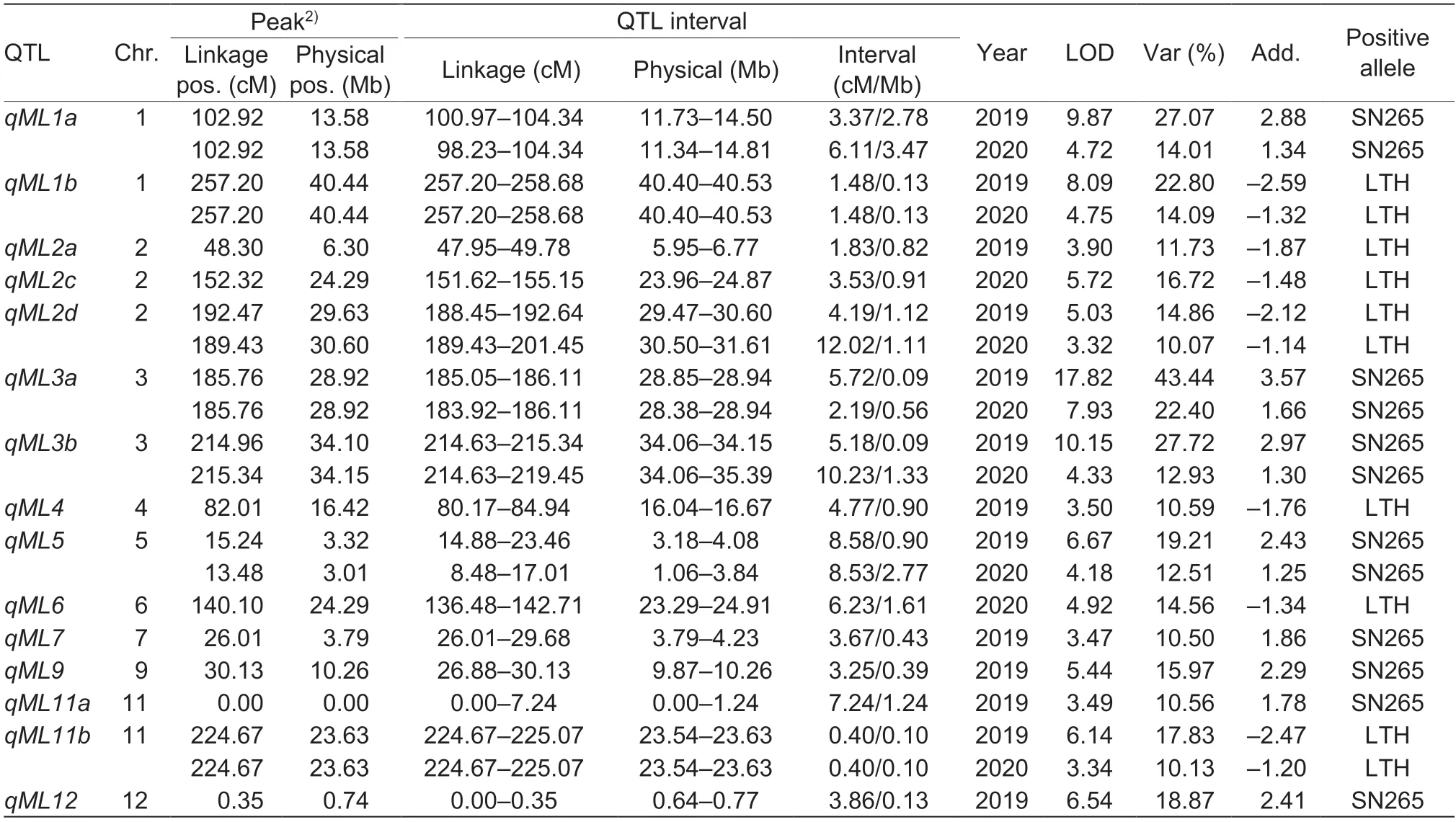
Table 2 Putative QTLs controlling mesocotyl length identified in the Lijiangxintuanheigu (LTH)/Shennong 265 (SN265) recombinant inbred lines via the interval mapping method1)
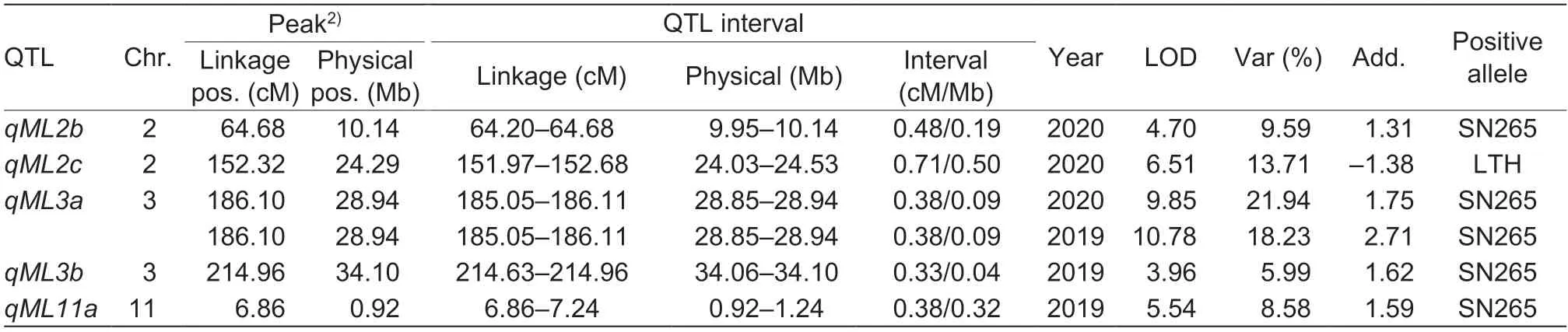
Table 3 Putative QTLs controlling mesocotyl length identified in the Lijiangxintuanheigu (LTH)/Shennong 265 (SN265) recombinant inbred lines via the composite interval mapping method1)
A total of 16 QTLs were identified in the two years.Seven QTLs,qML1a,qML1b,qML2d,qML3a,qML3b,qML5,andqML11b,were redetected in both yearsviathe interval mapping method.Only one major QTL,qML3a,was reidentified in the two yearsviathe composite interval mapping method.Four QTLs (qML2c,qML3a,qML3b,andqML11a) were found in different QTL mapping methods.qML2a,qML2b,qML4,qML6,qML7,andqML9were detected only once per year in this study.
3.4.Annotation of genes for each quantitative trait locus interval
The confidence interval of each QTL was assured by the smallest interval from the results of the interval mapping method and the composite interval mapping method(Tables 2-4).The QTLqML1ahad the largest confidence physical interval of 2.78 Mb with 413 putative candidate genes annotatedviathe Rice Genome Annotation Project database (Table 4;Appendix A).The physical interval ofqML1bwas 126.02 kb with 15 putative candidate genes(Table 4;Appendix B).A total of 124 genes (8 116.91 kb),28 genes (190.22 kb),73 genes (500.35 kb),and 21 genes (102.62 kb) were found in the physical confidence intervals ofqML2a,qML2b,qML2c,andqML2don chromosome 2,respectively (Table 4;Appendices CF).The major QTLsqML3aandqML3bhad 13 putative candidate genes with a confidence interval of 88.18 kb and 10 genes with a confidence interval of 37.69 kb,respectively (Table 4;Appendices G and H).The putative candidate genes of theqML4confidence interval andqML5confidence interval numbered 97 and 96,respectively (Table 4;Appendices I and J).qML6had the second-largest confidence physical interval of 1.61 Mb and had 227 putative candidate genes (Table 4;Appendix K).The 434.96 kb confidence physical interval ofqML7included 65 genes (Table 4;Appendix L).The 389.97 kb confidence physical interval ofqML9had 56 putative candidate genes (Table 4;Appendix M).The QTLsqML11aandqML11bhad 52 putative candidate genes with a confidence interval of 319.41 kb and 14 putative candidate genes with a confidence interval of 96.67 kb,respectively (Table 4;Appendices N and O).TheqML12had 24 putative candidate genes in its 129.01 kb physical interval (Table 4;Appendix P).The confidence intervals of the six QTLs (qML1b,qML2d,qML3a,qML3b,qML11b,andqML12) were smaller than 150 kb (Table 4).
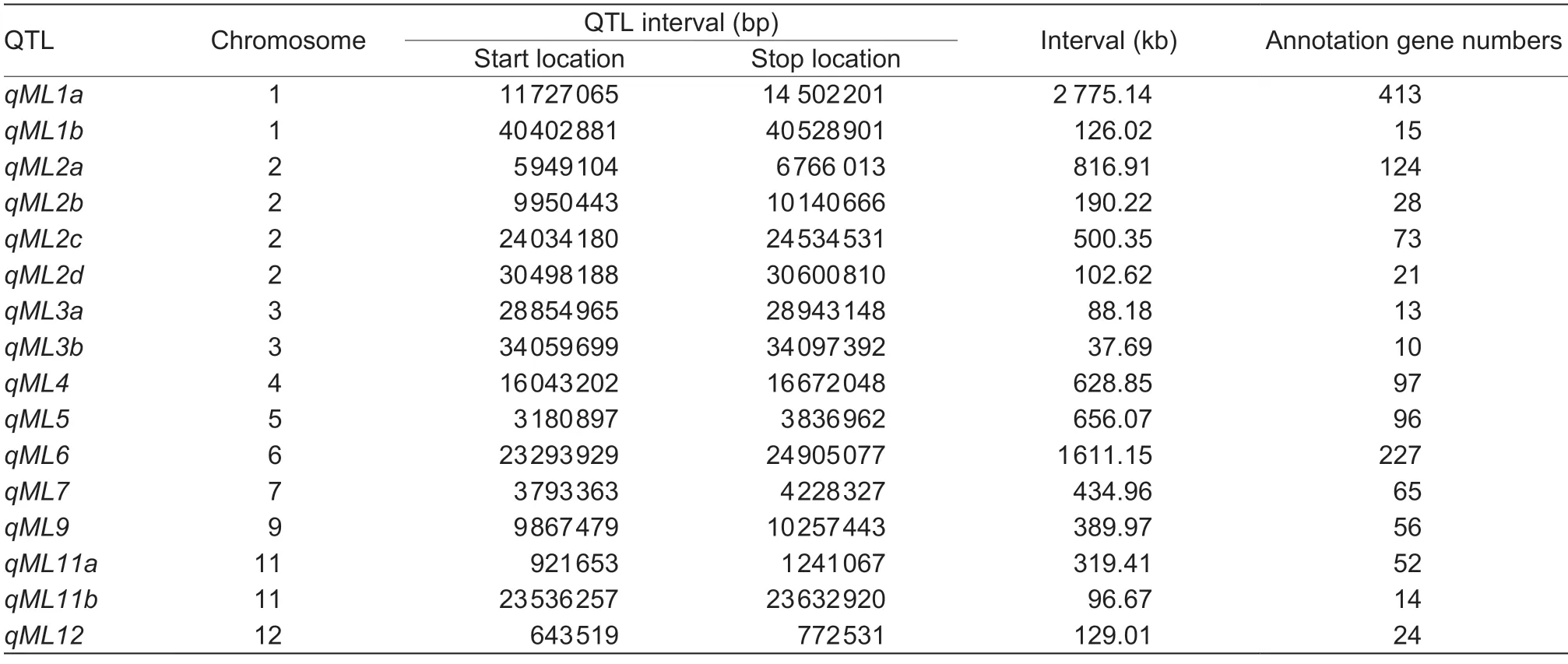
Table 4 The annotation gene numbers for each putative quantitative trait locus
3.5.Candidate genes and coding DNA sequence variations for qML3a
The major QTL,qML3a,associated with mesocotyl elongation was reidentifiedviatwo mapping methods (IM and CIM) in both years.It had the highest LOD value,explained the largest phenotypic variation,and covered a relatively small genetic interval (88.18 kb) (Fig.4-A;Table 2).Therefore,theqML3aQTL interval was further and more thoroughly analyzed in order to identify novel candidate genes controlling mesocotyl elongation.There were 13 putative candidate genes identified within theqML3ainterval (LOC_Os03g50540,LOC_Os03g50550,LOC_Os03g50560,LOC_Os03g50570,LOC_Os03g50580,LOC_Os03g50590,LOC_Os03g50600,LOC_Os03g50610,LOC_Os03g50620,LOC_Os03g50630,LOC_Os03g50644,LOC_Os03g50660,andLOC_Os03g50670) in the confidence interval ofqML3a(Fig.4-B;Appendix G).LOC_Os03g50540encodes a 2Fe-2S iron-sulfur cluster-binding domain-containing protein;LOC_Os03g50550encodes a mitogen-activated protein kinase kinase (MPKK);LOC_Os03g50560encodes an RNA recognition motif-containing protein;LOC_Os03g50570,LOC_Os03g50580,LOC_Os03g50590,LOC_Os03g50600,LOC_Os03g50610,LOC_Os03g50630,andLOC_Os03g50644all encode expressed proteins;LOC_Os03g50620encodes an ATP-binding protein;LOC_Os03g50660andLOC_Os03g50670encode a transposon protein and a retrotransposon protein,respectively.
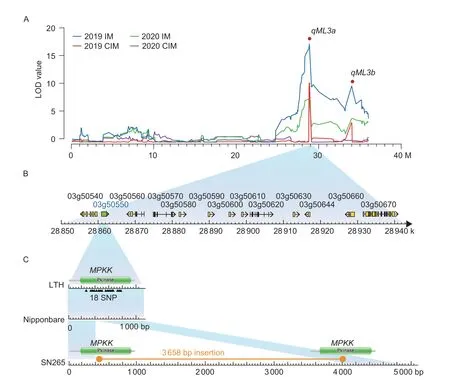
Fig.4 QTL location,candidate gene,and DNA sequence variations for qML3a.A,plots of the LOD values of the QTLs for mesocotyl elongation on chromosome 3 detected via the interval mapping (IM) method and composite interval mapping (CIM) method.B,the 13 putative candidate genes in the confidence interval of qML3a.C,the genomic DNA sequence variations of LOC_Os03g50550 (the high confidence candidate gene for qML3a) between Shennong 265 (SN265),Lijiangxintuanheigu (LTH),and Nipponbare.M,million base pairs;k,kilo base pairs;SNP,single nucleotide polymorphism.
Analyses of re-sequencing data in the delimited regions of each candidate gene were carried out to detect the genomic DNA polymorphisms between SN265 and LTH.A total of 47 SNPs were found in the exon regions ofLOC_Os03g50540,LOC_Os03g50550,LOC_Os03g50570,LOC_Os03g50580,LOC_Os03g50600,LOC_Os03g50610,LOC_Os03g50620,LOC_Os03g50644,LOC_Os03g50660,andLOC_Os03g50670(Table 5).LOC_Os03g50540hadtwo SNPs in exon 1 (28 857 722) and exon 4 (28 859 910).A total of 18 SNPs were identified in the exon regions ofLOC_Os03g50550.LOC_Os03g50570had two SNPs in exon 1;LOC_Os03g50580,LOC_Os03g50600,LOC_Os03g50620,andLOC_Os03g50644had two SNPs in their exon regions;only one SNP was identified in the exon region ofLOC_Os03g50610;12 SNPs were found in exon 1 ofLOC_Os03g50660;four SNPs were found in exon 2 ofLOC_Os03g50670.
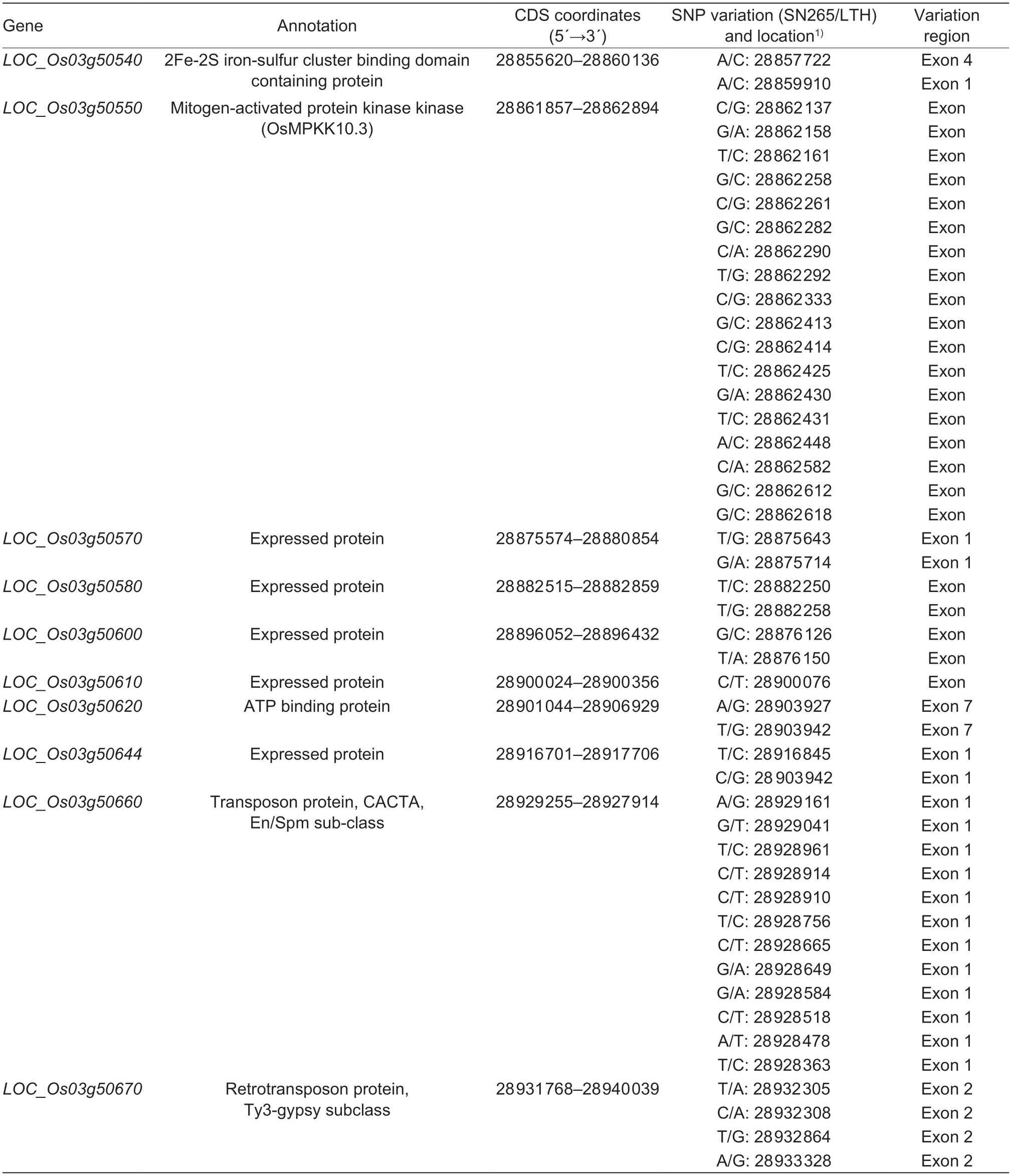
Table 5 SNPs detected in the putative candidate genes of the QTL qML3a controlling mesocotyl elongation in rice
To ensure the annotation of high-confidence candidate genes for the major QTLqML3a,the whole-genome sequence of these genes in the QTL region from SN265(long mesocotyl) was further compared with Nipponbare(short mesocotyl).No variations were found in the gene sequences ofLOC_Os03g50560,LOC_Os03g50580,LOC_Os03g50590,LOC_Os03g50600,LOC_Os03g50610,LOC_Os03g50620,LOC_Os03g50630,andLOC_Os03g50660between SN265 and Nipponbare.One SNP was detected in the 5´UTR ofLOC_Os03g50540.Two SNPs and one SNP were found in the intron regions ofLOC_Os03g50570andLOC_Os03g50670,respectively.One SNP (28 917 018) was identified in the exon region ofLOC_Os03g50644,but this SNP was not found between SN265 and LTH.Although none of the SNPs were found inLOC_Os03g50550between SN265 and Nipponbare,a large insertion of 3 658 bp was identified in the exon ofLOC_Os03g50550in the SN265 genome.This insertion was not found in LTH (Fig.4-C).All these DNA sequence variations among SN265,LTH,and Nipponbare indicated thatLOC_Os03g50550was the high-confidence forqML3a.TheLOC_Os03g50550locus from SN265 encoded two protein kinases,whereas this locus from Nipponbare encoded one protein kinase according to a search in the Pfam database (http://pfam.xfam.org/).SeveralLOC_Os03g50550homologs were annotated as mitogen-activated protein kinase kinase(MPKK) in the NCBI database (Fig.4-C).
3.6.The phenotypic verification of qML3a and expression analysis of LOC_Os03g50550
TwoqML3a-NILs with 98.53% genome similarity were used to perform the phenotypic verification in two years.A 756.8-kb(28 186 391-28 943 148 bp) SN265 genome segment on chromosome 3 was different between the two NILs (Fig.5-A).NIL-qML3ahas longer mesocotyl as SN265,and NIL-qml3ahas shorter mesocotyl as LTH (Fig.5-B).The mesocotyl length (mean±SD) of NIL-qML3awas (17.00±4.02) mm in 2019 and (14.92±3.01) mm in 2020,whereas the mesocotyl length of NIL-qml3awas (6.96±3.25) mm in 2019 and (7.96±3.19) mm in 2020 (Fig.5-C).The mesocotyl length of NIL-qML3awas (6.33±1.30) mm in the soil culture method,whereas the mesocotyl length of NILqml3awas (2.11±0.78) mm (Fig.5-D).This information verified thatqML3awas the major QTL controlling mesocotyl elongation in SN265.To further investigate the gene expression levels ofLOC_Os03g50550,qRTPCR analyses were executed to evaluate the expression patterns ofLOC_Os03g50550in two parents and twoqML3a-NILs.The expression levels ofLOC_Os03g50550in mesocotyls of LTH and NIL-qml3awere significantly lower than in the mesocotyls of SN265 and NIL-qML3a(Fig.5-E),which indicates thatLOC_ Os03g50550gene is essential for rice mesocotyl elongation and is the strongest candidate gene for the major QTLqML3a.
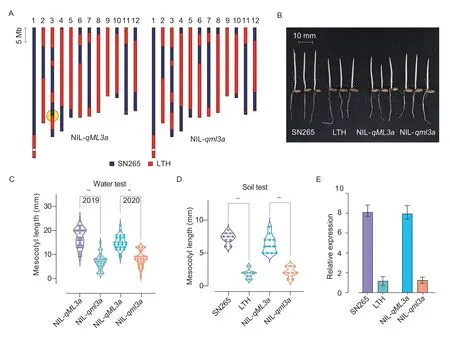
Fig.5 The genotype,mesocotyl phenotypic verification,and expression analysis of qML3a.A,the genotype of two qML3a-NILs(NIL-qML3a and NIL-qml3a).B,the mesocotyl phenotype of the two parents (Shennong 265 (SN265) and Lijiangxintuanheigu(LTH)) and two qML3a-NILs (NIL-qML3a and NIL-qml3a) in the soil culture method.C,the mesocotyl phenotype of the two qML3a-NILs (NIL-qML3a and NIL-qml3a) in the water culture method.D,the mesocotyl length of two parents and two qML3a-NILs in the soil culture method.E,the relative gene expression of the LOC_Os03g50550 gene between two parents and two qML3a-NILs.Bars mean SD (n=3).**,P<0.01,using Student’s t-test.
4.Discussion
In recent years,DSR has been urgently needed in many Asian countries to relieve the conflicts between resource shortages (such as labor,water,and other resource shortages) and high input costs of traditional transplanted rice (TPR) systems (Sagareet al.2020).However,the varieties currently used in DSR were developed for TPR.These TPR varieties could not exactly meet the requirements for DSR production,especially the requirements for early uniform emergence and early seedling establishment to achieve the potential yield of DSR (Kumar and Ladha 2011).Many studies have revealed that mesocotyl length is directly related to seedling emergence and that enhanced mesocotyl elongation ability is associated with better seedling emergence and establishment (Leeet al.2017;Sagareet al.2020;Zhanet al.2020).Therefore,the development of new cultivars with excellent mesocotyl elongation ability and discoveries of novel and key genes responsible for mesocotyl elongation are essential to ensure the application of new DSR varieties to achieve the maximal yield potentials.
4.1.Some QTLs for mesocotyl elongation in SN265 originated from its indica ancestor
Generally,weed rice has a longer mesocotyl thanindicarice,andindicarice has a longer mesocotyl thanjaponicarice (Liet al.2012).Compared toindicarice,the mesocotyl ofjaponicarice exhibits almost no elongation.In this study,SN265,the first released commercialjaponicasuper rice variety derived from the cross betweenjaponicaandindica,was selected since it displays a long mesocotyl character.The mesocotyl length (mean±SD)of SN265 was (17.64±6.91) mm in 2019 and (13.13±6.20)mm in 2020.A total of nine QTLs were identified with positive allele from SN265.The pedigree analysis shows that genes regulating mesocotyl length in ‘SN265’ might be derived from itsindicaancestor (Fig.6) (Huanget al.2010).The 4 696 bp genomic DNA fragment ofLOC_Os03g50550(the candidate gene for the major QTLqML3a) from SN265 was BLAST searched against the NCBI database and the Molecular Breeding Knowledgebase (http://mbkbase.org/rice),and the BLAST search results showed that more than 98% of sequence similarities were shared with the sixindicavarieties Zhenshan 97,Minghui 63,N22,9311,IR8,and Shuhui 498.These results definitely support the conclusion that the long mesocotyl gene is derived fromindicarice.
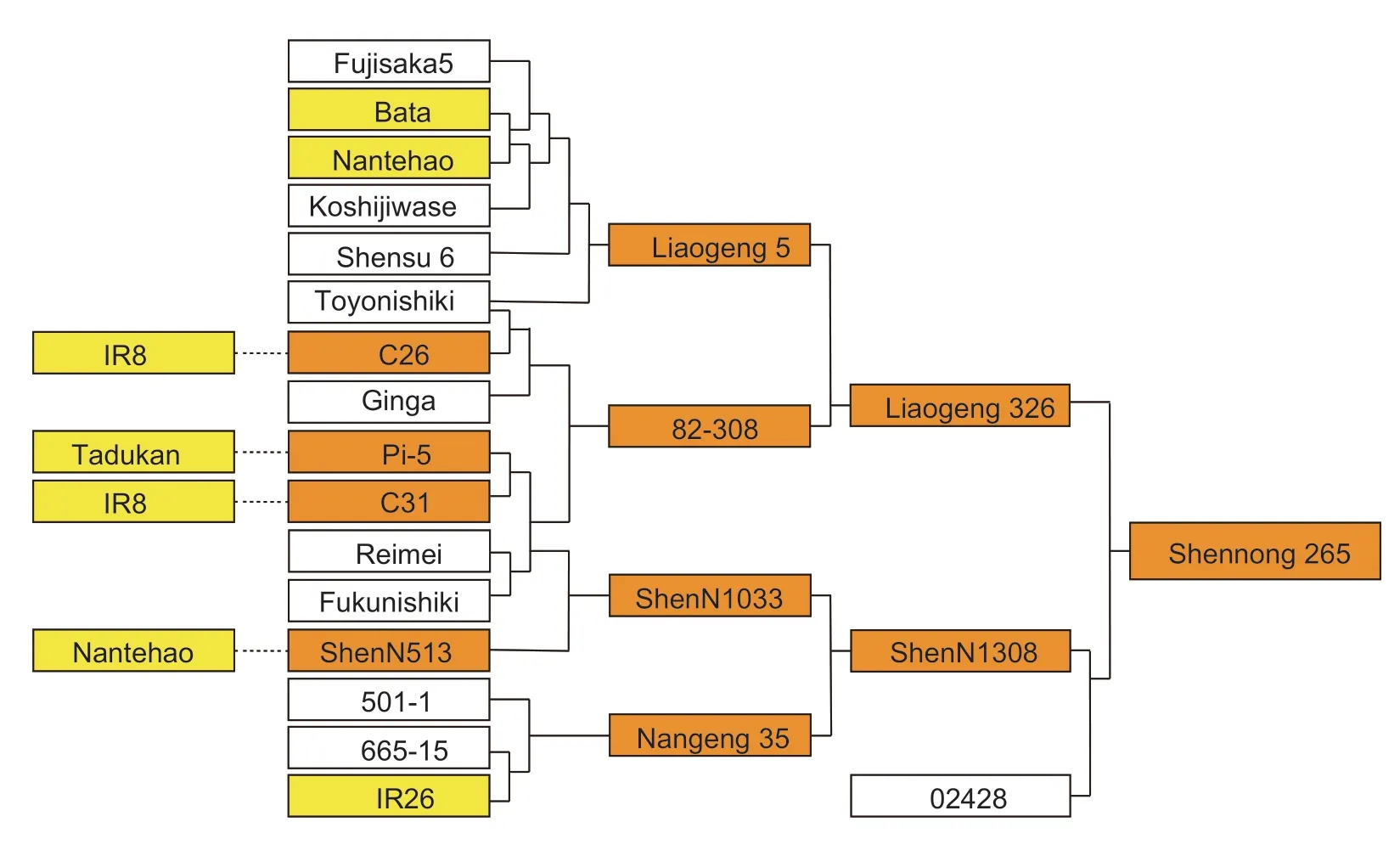
Fig.6 The pedigree of Shennong 265.Yellow is the indica ancestor and orange is the ancestor or parent containing indica consanguinity.
4.2.Resequencing linkage maps are an effective way to shrink the interval of QTLs
Advances in whole genome resequencing technology have provided abundant SNP markers for genetic mapping analysis,especially for closely related rice materials (Jianget al.2020b).Compared to traditional genetic maps,such as restriction fragment length polymorphism (RFLP) marker maps and simple sequence repeats (SSR) marker maps,re-sequenced bin maps have a higher marker density and higher accuracy (Cai and Morishima 2002;Huanget al.2010;Leeet al.2012b;Luet al.2016;Jianget al.2020b;Liuet al.2020).The bin map constructed by re-sequenced SNPs could provide precise physical locations to further narrow the interval of QTLs.An F6population derived from the same cross combination of LTH/SN265 was previously used for QTL mapping for mesocotyl elongation (Huanget al.2010).The interval ofqML2was narrowed down approximately 6-fold from 4.83 (2.88-7.71) Mb to 0.82(5.95-6.77) Mb and remapped asqML2a.The interval ofqML3decreased approximately 40-fold from 3.58 (27.40-30.98) Mb to 0.09 (28.85-28.94) Mb and was remapped asqML3a.The interval ofqML11shrank approximately 18-fold from 5.98 (0.03-6.01) Mb to 0.32 (0.92-1.24) Mb and was remapped asqML11a.In addition,the QTL detection ability using resequencing maps is also significantly enhanced compared with traditional molecular marker maps.We previously detected five QTLs for mesocotyl length using traditional molecular marker maps but identified 16 QTLs in this study using resequencing linkage maps.
4.3.Comparison of the locations of mesocotyl elongation QTLs in this study and in previous studies
A large number of QTLs controlling mesocotyl elongation have been detected using different mapping populations,including RILs (Huanget al.2010),Backcross recombinant inbred lines (BILs) (Leeet al.2012b),chromosome segment substitution lines (CSSLs),double haploid (DH) populations (Caoet al.2002),and GWAS populations.These QTLs are located on all 12 rice chromosomes (Zhanet al.2020).By comparing the positions of the QTLs,10 of the previously identified QTL regions for mesocotyl elongation were found in this study,as summarized in Table 6.qML1awas detected previously as a QTL namedqML1(Liuet al.2020),qMel-1(Leeet al.2012a),andMSL1(Cai and Morishima 2002).The QTL location ofqML1bis very close toqME1(Wuet al.2015),qMel-1(Leeet al.2012b),andGWAS locusseq-rs576 (Luet al.2016) and includes two cloned mesocotyl associated genes,sd1(Lv 2020) andGY1(Xionget al.2017).qML2awas also detected by Huanget al.(2010) asqML2and Liuet al.(2020) asqML2a.qML3awas identified as a major QTL in many different mapping populations (Cai and Morishima 2002;Huanget al.2010;Leeet al.2012a,b;Zhaoet al.2018).qML3b,qML4,andqML5were identified by Cai and Morishima(2002) asMSL3-3,MSL4,andMSL5,respectively.qML6was found near the GWAS locusqML6by Wuet al.(2015).qML9was a stable QTL that could be detected in different populations (Cai and Morishima 2002;Leeet al.2012b;Luet al.2016).The previously detected locusqML11was reidentified in this study by using the same population(Huanget al.2010).A total of six QTLs (qML2b,qML2c,qML2d,qML7,qML11b,andqML12) were found for the first time in this study.The above results reflect not only the accuracy of this study but also the complexity of mesocotyl elongation.Moreover,this study shows that mesocotyl elongation ability has strong genetic stability.
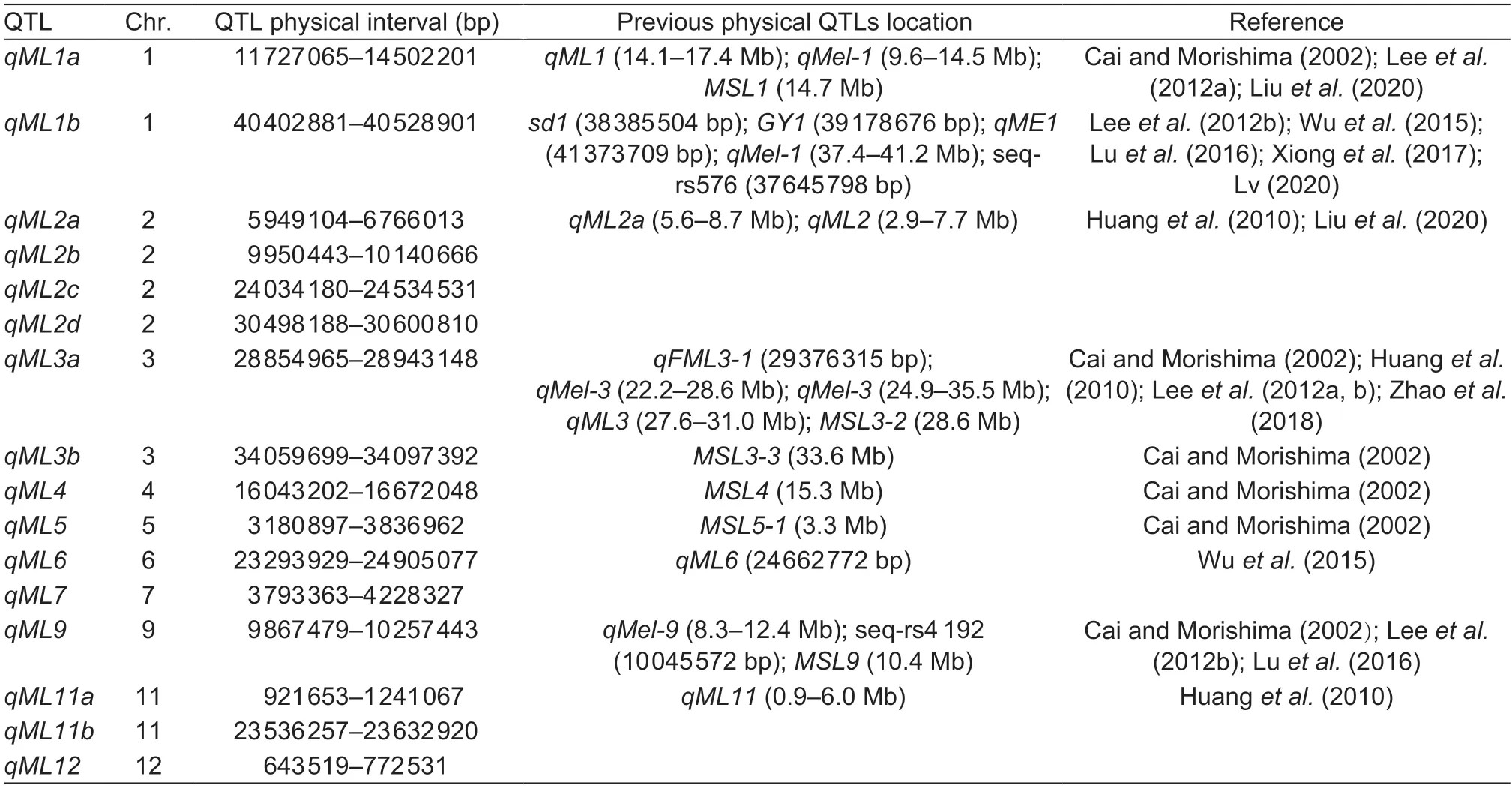
Table 6 Comparison of QTLs for mesocotyl elongation to those detected in prior studies
4.4.LOC_Os03g50550 encoding an MPKK is the high-confidence candidate gene for qML3a
It is well known that mitogen-activated protein kinase(MAPK) modules play key roles in the signal transmission pathway mediated by auxin,ethylene,jasmonic acid,and abscisic acid (Jagodziket al.2018).Recent studies have shown that theAtYODA-AtMKK4/5-AtMPK3/6module is involved in hypocotyl development inArabidopsis thalianathrough brassinosteroid (BR) biosynthesis (Leet al.2014).Recently,a conserved GSK3-like kinase was identified tobe involved in brassinosteroid signaling,and it determines the natural variation in mesocotyl elongation (Sunet al.2018).The signaling transcription of ethylene,another important phytohormone controlling mesocotyl elongation,was also found to be regulated by theMKK9-MPK3/MPK6cascade inArabidopsis(Yooet al.2008).The above information supports thatLOC_Os03g50550encoding a mitogen-activated protein kinase kinase (MPKK) is the high-confidence candidate gene responsible for the major QTLqML3a.
5.Conclusion
This study identified a total of 16 QTLs for mesocotyl length on chromosomes 1(2),2(4),3(2),4,5,6,7,9,11(2),and 12.Thirteen QTLs for mesocotyl elongation were identified in 2019,and nine QTLs controlling mesocotyl elongation were identified in 2020.Seven QTLs were reproducibly detected in both years,includingqML1a,qML1b,qML2d,qML3a,qML3b,qML5,andqML11b viathe interval mapping method.OnlyqML3awas reidentified in both yearsviathe composite interval mapping method.Thirteen predicted genes covering the 88.18 kb genetic interval ofqML3awere comprehensively analyzed.Finally,coding DNA sequence variations among SN265,LTH,and Nipponbare and the qRT-PCR results indicated thatLOC_Os03g50550is the strongest candidate gene responsible for theqML3aQTL and encodes a mitogen-activated protein kinase (MPKK).These results further strengthen our knowledge about the genetic mechanism controlling mesocotyl elongation ability in rice and help to accelerate rice breeding programs for DSR variety development.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Heilongjiang Province,China(LH2020C098),the Fundamental Research Funds for the Research Institutes of Heilongjiang Province,China(CZKYF2020A001),the National Key Research and Development Program of China (2016YFD0300104),and the Heilongjiang Province Agricultural Science and Technology Innovation Project,China (2020JCQN001,2019JJPY007,2020FJZX049,2021QKPY009,2021CQJC003).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- OsMas1,a novel maspardin protein gene,confers tolerance to salt and drought stresses by regulating ABA signaling in rice
- Marker-assisted selection to pyramid Fusarium head blight resistance loci Fhb1 and Fhb2 in the high-quality soft wheat cultivar Yangmai 15
- Identification,evolution,expression and protein interaction analysis of genes encoding B-box zinc-finger proteins in maize
- Characterization of transgenic wheat lines expressing maize ABP7 involved in kernel development
- Increasing the appropriate seedling density for higher yield in dry direct-seeded rice sown by a multifunctional seeder after wheatstraw return
- Nitrogen management improves lodging resistance and production in maize (Zea mays L.) at a high plant density