P-HPLC法同时测定不同产地不同采收期墨旱莲中6种成分的含量
2023-02-02南敏伦司学玲司英奇张瀚水龚本义赵昱玮赫玉芳
南敏伦,尹 涵,司学玲,司英奇,张瀚水,龚本义,赵昱玮,赫玉芳*
(1. 吉林省中医药科学院,长春 130012;2. 吉林农业大学,长春 130118;3. 修正药业集团长春高新制药有限公司,长春 130012;4. 吉林省奥普金钻技术检测服务有限公司,长春 130507;5. 长春工业大学,长春 130021;6.长春中医药大学,长春 130117)
墨旱莲为菊科植物鳢肠(Eclipta prostrataL.)的干燥地上部分[1],又名旱莲草、水旱莲、猪牙草、莲子草、黑墨草。具有滋补肝肾、乌须固齿、凉血止血的功效[1],广泛分布于广东、广西、江苏、河南、福建等地。墨旱莲化学成分包含黄酮、香豆素、三萜皂苷、有机酸类、噻吩及挥发油[2],目前《中国药典》2020年版仅以蟛蜞菊内酯单一成分的含量墨旱莲药材的质量进行控制[1,3],不能全面整体反映墨旱莲药材的质量,并且对产地及采收期的研究相对较少。本研究针对以上情况,采集6个产地18批不同采收期的墨旱莲药材进行系统研究,采用HPLC法对墨旱莲中含有的主要成分咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯进行含量测定[4-8],初步揭示了不同产地、不同采收期墨旱莲药材中各成分含量有差异,为严格把控墨旱莲药材的质量,资源合理应用,以及该药材更深入的开发研究提供依据。
1 仪器与试药
1.1 仪器
Agilent 1260(美国安捷伦公司);BS 124S电子天平(赛得利斯);MD-400KDE型台式高功率数控超声波清洗器(苏州美达超声仪器厂)。
1.2 试药
咖啡酸对照品(中国食品药品检定研究院,批号:110885-201703, 纯度:99.7%); 木犀草苷对照品(中国食品药品检定研究院,批号:111720-201609,纯度:94.9%);异绿原酸B对照品(中国食品药品检定研究院,批号:111782-201807,纯度:94.3%);异绿原酸C对照品(中国食品药品检定研究院,批号:111894-202103,纯度:95.2%);木犀草素对照品(中国食品药品检定研究院,批号:111520-202005,纯度:99.6%);蟛蜞菊内酯对照品(中国食品药品检定研究院,批号:111885-202105,纯度:97.2%);水为娃哈哈纯净水;其他试剂均为分析纯;实验所用墨旱莲药材于2020年4-6月采自不同产地,由吉林省中医药科学院赵全成研究员鉴定均为真品。
2 方法与结果
2.1 色谱条件
色谱柱为Agilent C18(250 mm × 4.6 mm,5 um);流动相A相为0.3%甲酸水,B相为乙腈溶液,梯度洗脱:0~10 min A由90%降至85%;10~35 min A由85%降至75%;35~50 min A由75%降至65%;50~70 min A由60%降至30%;70~80 min A由30%降至20%;流速为0.9 mL·min-1;检测波长为340 nm;柱温为30 ℃;进样量10 μL。对照品及供试品溶液色谱图见图1。
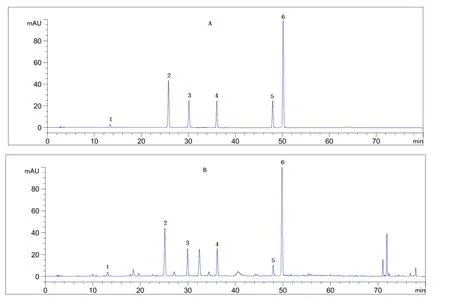
图1 六种对照品(A)及样品(B)高效液相色谱图
2.2 对照品溶液的制备
精密称取对照品咖啡酸10.07 mg、木犀草苷10.76 mg、异绿原酸B 10.85 mg、异绿原酸C 10.46 mg、木犀草素10.17 mg和蟛蜞菊内酯10.36 mg,分别置25 mL容量瓶中,用甲醇溶解并定容至刻度,分别得到6种对照品储备液;分别精密量取咖啡酸储备液0.25 mL,木犀草苷储备液0.5 mL,异绿原酸B储备液1.0 mL,异绿原酸C储备液1.0 mL,木犀草素储备液1.0 mL,蟛蜞菊内酯储备液5 mL置同一10 mL容量瓶中,用甲醇稀释至刻度,制备得到每毫升含咖啡酸0.010 04 mg、木犀草苷0.020 42 mg、异绿原酸B 0.040 93 mg、异绿原酸C 0.039 83 mg、木犀草素0.040 52 mg、蟛蜞菊内酯0.201 40 mg的对照品混合溶液。
2.3 供试品溶液的制备
取墨旱莲药材粉末(过60目筛)1.0 g,精密称定,置25 mL容量瓶中,加入约80%甲醇20 mL,超声处理60 min,冷却,用甲醇定容至刻度,摇匀,滤过(0.45 mm),即得。
2.4 线性关系考察
上述对照品混合溶液分别按照2 µL、5 µL、10 µL、15 µL、20 µL 进样测定,以峰面积Y为纵坐标,进样量X(μg)为横坐标,绘制标准曲线,计算回归方程(见表1)。说明咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯在线性范围内线性良好。

表1 6个对照品的标准曲线 μg
2.5 精密度试验
取供试品溶液(1号样),连续进样6次,记录峰面积;咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯峰面积的RSD分别为0.85%、0.64%、0.38%、0.72%、0.66%、0.76%,表明仪器的精密度良好。
2.6 重复性试验
取同一批供试品(1号样)6份,按制备方法,制备得到供试品溶液,按色谱条件进样测定,记录峰面积,计算含量。计算结果表明,咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯含量的RSD分别为0.82%、0.64%、0.59%、1.01%、1.12%、0.75%,表明该方法重复性良好。
2.7 稳定性试验
取供试品溶液(1号样),分别于0 h、2 h、4 h、8 h、12 h、24 h进行测定,记录峰面积,计算含量,实验结果表明咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯含量的RSD分别为0.48%、0.55%、0.67%、0.84%、0.91%、0.64%,表明供试品溶液在24 h内稳定。
2.8 加样回收率试验
取已知含量的供试品(1号样)6份,各0.5 g,精密称定,分别加入适量的对照品,按供试品制备方法制备,按照色谱条件测定,记录峰面积,计算回收率(见表2)。回收率结果表明,6种化合物的加样回收率良好。
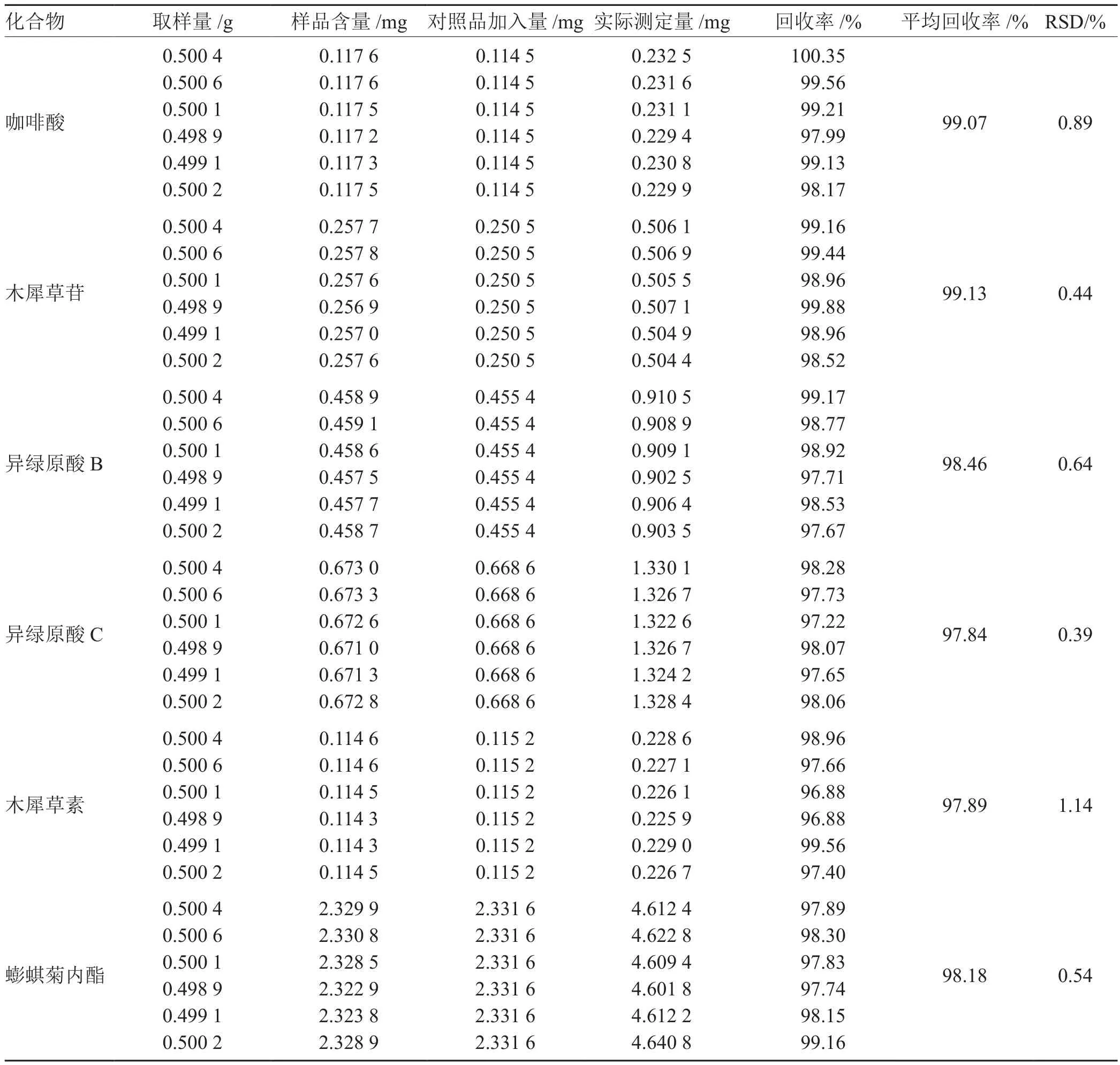
表2 加样回收率实验结果
2.9 样品含量测定
取不同产地和不同采收期的墨旱莲样品,制备供试品溶液,测定并计算咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯的含量(见表3)。
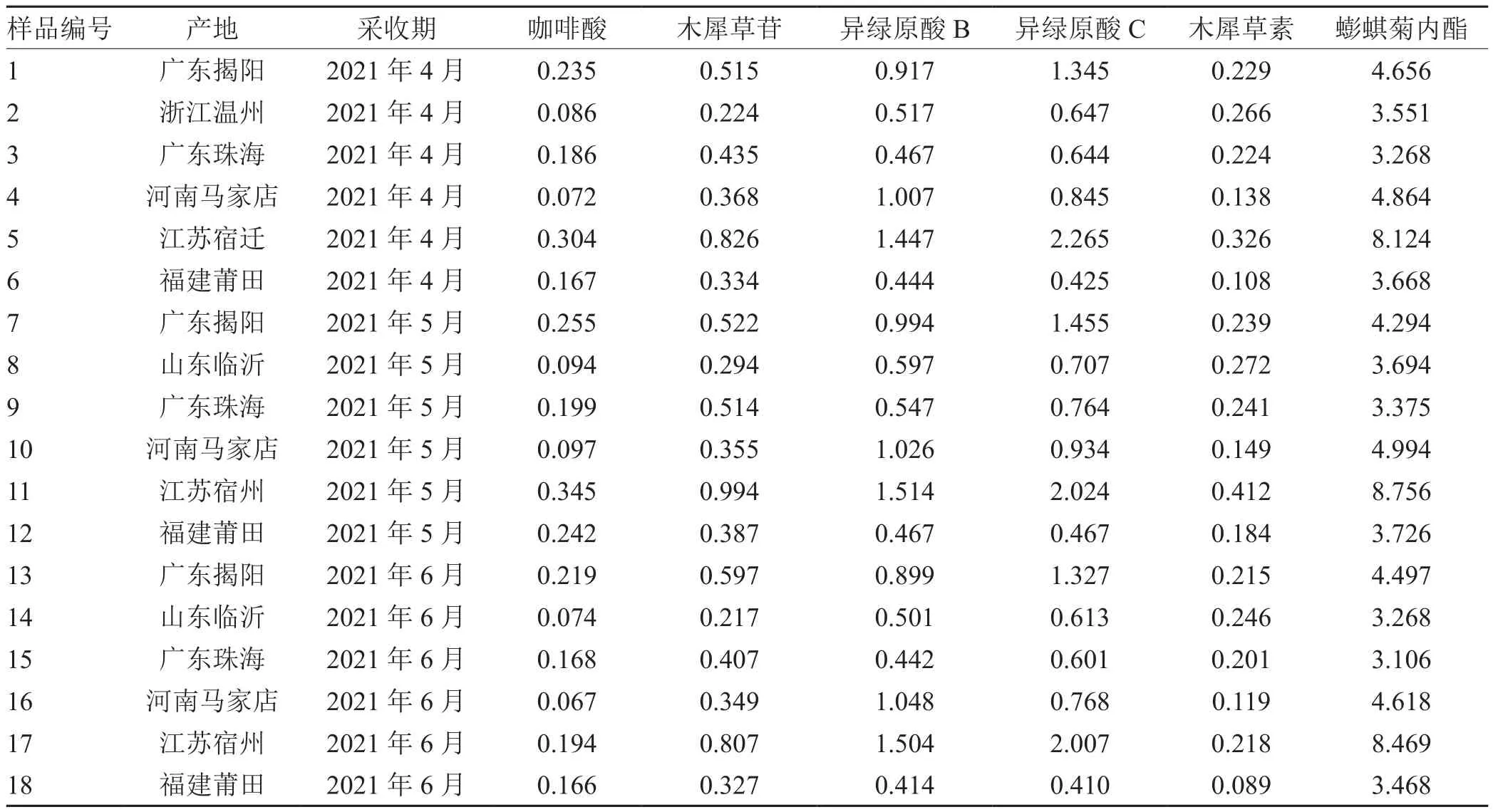
表3 不同产地不同采收期墨旱莲中6种成分的含量测定(n= 3) mg·g-1
3 讨论
由于墨旱莲药材中化学成分含有黄酮类、有机酸类、噻吩类等[2],化学成分具有多样性,只采用甲醇-水、乙腈-水系统不能将有效成分进行有效分离。经过多次试验,以乙腈-0.3%甲酸水溶液为流动相,采用梯度洗脱,待测样中咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯达到基线分离;采用340 nm为检测波长,目标峰和杂质峰可以达到完全分离;由于咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯等6种化合物剂型差别较大,为了能使各化合物均能完全提取出来,选用不同浓度乙醇及不同浓度甲醇对提取溶剂进行考察,结果表明80%甲醇提取效果更好;同时考察了超声和回流两种提取方法,实验结果表明,两种提取方法各成分提取效率基本一致,由于超声提取方法操作简单,故此选择超声提取;提取时间考察结果显示,在60 min内各目标化合物均可以完全提取。故本实验以80%甲醇为提取溶剂,提取时间为60 min。
本实验对不同产地和不同采收期的墨旱莲药材中的咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯进行含量测定研究,通过实验看出,不同产地的咖啡酸、木犀草苷、异绿原酸B、异绿原酸C、木犀草素、蟛蜞菊内酯含量具有明显的不同,同一采收期药材江苏宿迁含量最高。不同采收期实验结果表明,同一产地5月份采收的墨旱莲6种成分的含量最高。由于这几个地区,墨旱莲开花日期基本均在5月份,墨旱莲在花开始开放还未完全开放时有效成分的含量最高,为墨旱莲的采收期的确定奠定基础。
本研究采用建立的含量测定方法对含墨旱莲药材中的6种成分进行含量测定研究,实验结果表明,本方法适合于墨旱莲药材的含量测定,其分离度、耐用性等均符合规定,可以为墨旱莲药材的质量控制研究奠定基础。
