毕赤酵母多基因组装系统的构建及其在2-苯乙醇合成中的应用
2023-02-02陈晓瑞战春君白仲虎杨艳坤
陈晓瑞,战春君,白仲虎,杨艳坤*
1(江南大学 生物工程学院,江苏 无锡,214122)2(江南大学,粮食发酵与食品生物制造国家工程研究中心,江苏 无锡,214122)
巴斯德毕赤酵母(Komagataellaphaffii)是一种甲基营养酵母,具有高密度发酵、副产物少、甲醇利用等特点,被广泛应用于外源蛋白的生产[1]。随着基因操作工具的不断发展,毕赤酵母也开始被用于代谢产物的合成[2],如脂肪醇的生产[2-3]。构建细胞工厂常需要同时过表达多个不同基因,Gibson组装[4]、golden gate克隆[5]、Gateway等克隆技术是构建多基因表达载体的有效方法。其中golden gate克隆技术主要利用二型内切酶切割位点在识别位点之外的特点,只需在片段两侧引入很短的序列即可实现无痕连接[6]。基于这种方法,PRIELHOFER等[7]建立了GoldenPics克隆方法,实现了8个表达盒的组装。通过设计不同的接头序列,这种方法可以实现多元件的顺序组装。GoldenPics是专用于毕赤酵母基因改造的试剂盒,相比于另一种酵母多元件质粒构建试剂盒Yeast Toolkit(YTK)[8],其连接各元件的接头序列被直接设计在骨架质粒上,而不是作为一个独立元件,避免了YTK中易产生错误连接的问题。但GoldenPics所构建的质粒需要经过菌落PCR验证,不如YTK中替换荧光蛋白的筛选方式直观。另外GoldenPics无法回收筛选标记,构建产生的菌株具有抗性,不够安全,不适合发酵工业生产需要,且无法实现游离表达,目的基因只能整合到基因组中。
针对以上问题,本研究设计了用于抗性回收及游离表达的元件,构建了一种高效的毕赤酵母多基因组装系统(multigene assembly system,MGAS),可灵活替换目的基因的启动子、终止子、整合位点及表达方式。2-苯乙醇(2-phenylethanol,2-PE)是一种具有玫瑰香气的高级芳香醇,具有很高的商业价值[9]。其生物合成途径较长(图1),调控机制复杂[10-12]。本研究利用MGAS,游离表达筛选了不同来源的关键酶并整合表达5个基因,最终重组菌株PE-3的2-PE产量达到了408.4 mg/L,相比出发菌株提高了10倍以上。该研究为毕赤酵母代谢改造提供了一种高效的分子工具,也为毕赤酵母合成高价值化学品提供参考。
1 材料与方法
1.1 菌株与质粒
实验所用菌株及质粒见附表1(https://kns.cnki.net/kcms/detail/11.1802.ts.20220512.1050.004.html,下同)。
1.2 培养基及试剂
1.2.1 培养基
LB培养基(g/L):酵母提取物5,胰蛋白胨10,NaCl 10,配制固体培养基时加入琼脂粉20。YPD培养基(g/L):酵母提取物10,胰蛋白胨20,葡萄糖20,配制固体培养基时加入琼脂粉20。YPM(yeast extract peptone methanol medium)培养基:0.5%(体积分数)甲醇,酵母提取物10 g/L,胰蛋白胨20 g/L。MD(minimal dextrose medium)培养基(g/L):葡萄糖20,YNB 13.4,配制固体培养基时加入琼脂粉20。BMGY(buffered glycerol-complex medium)培养基:酵母提取物10 g/L,胰蛋白胨20 g/L,甘油2.5 g/L,YNB 13.4 g/L,100 mmol/L磷酸钾缓冲液(pH=6.0)。BMM(buffered methanol medium)培养基:0.5%(体积分数)甲醇,YNB 13.4 g/L,100 mmol/L磷酸钾缓冲液(pH=6.0)。

图1 毕赤酵母从头合成2-PE途径示意图Fig.1 De novo synthesis of 2-PE in Komagataella phaffii
1.2.2 试剂
所有内切酶均购自Thermo Fisher Scientific;MultiF Seamless Assembly Mix购自ABclonal;Ligation mix购自Takara;PCR Mix及回收胶、纯化、质粒提取所用试剂盒均购自康为世纪;2-PE标准品购自Sigma;其余试剂均购自国药集团化学试剂有限公司。本文所用引物如附表2所示均合成自苏州金唯智生物科技有限公司。
1.3 摇瓶培养条件及抗性消除方法
1.3.1 摇瓶培养
将50 μL 菌液接种5 mL YPD培养基,30 ℃,200 r/min下过夜培养。之后按终浓度OD600=0.1接种至50 mL BMGY培养基,待OD600值达到4~6后收集菌液,5 000 r/min离心5 min,弃上清液保留菌体,转接至50 mL BMM培养基中进行发酵。每隔24 h补加终体积分数为0.5%的甲醇并取样测定生物量和2-PE浓度。
1.3.2 抗性消除方法
将50 μL菌液接种5 mL YPM培养基,30 ℃,200 r/min下培养24 h,划线至YPD平板。30 ℃培养48 h,挑取单菌落分别接种至YPD及含0.1 mg/L博来霉素的YPD(YPDZ)培养基中,30 ℃,200 r/min下培养36 h,只能在YPD中生长的菌落即为抗性消除菌。
1.4 测定方法
样品处理方法:取发酵液1 mL,12 000 r/min离心5 min后取上清液,加入等量无水乙醇,混匀后12 000 r/min离心5 min,取上清液进行HPLC测定。
2-PE使用HPLC C18色谱柱(Shim-pack GIST,5 μm,150 mm×4.6 mm,日本岛津)进行检测,HPLC程序为:柱温30 ℃,流速1 mL/ min,检测波长214 nm,A相为含有1%(体积分数)乙腈的20 mmol/L KH2PO4溶液,B相为乙腈,0~6 min B相浓度由0%升至10%,6~25 min B相浓度由10%升至60%,25~26 min B相浓度由60%降至0%,保持至28 min。2-PE出峰时间为16.4 min。
2 结果与分析
2.1 毕赤酵母多基因表达系统的构建
为构建毕赤酵母多基因表达系统,首先构建了一系列带有接头序列的骨架质粒,之后在此基础上插入了启动子、终止子、整合位点、CEN/ARS序列、Cre-lox序列等一系列元件。
2.1.1 骨架质粒的构建
通过将pYTK001的BsmB I酶切位点替换为BsaI/BbsI酶切位点并引入GoldenPics中的接头序列构建了Bb1系列的骨架质粒,接头序列及质粒图谱如图2所示。以pYTK001为模板,利用引物对Bb1-12-Fs1-F、Bb1-12-Fs2-R和Bb1-12-Fs2-F、Bb1-12-Fs1-R进行PCR,得到片段Bb1-12-F1和Bb1-12-F2。通过同源重组连接两片段,得到质粒Bb1-12。质粒Bb1-23、Bb1-34使用了相同的构建方法。Bb3系列的骨架质粒构建方法与Bb1相同,区别为构建Bb3-AD使用的引物对为Bb3-FsD-F、Bb3-AD-FsD-R和Bb3-FsA-R、Bb3-AD-FsD-R,Bb3-AH使用的引物对为Bb3-FsA-F、Bb3-AH-FsH-R和Bb3-FsA-R、Bb3-AH-FsH-R。
通过将pYTK095的BsaI酶切位点替换为BbsI/BsaI酶切位点并引入GoldenPics中的接头序列构建了Bb2系列的骨架质粒。以pYTK095为模板,利用引物对Bb2-AB-FsA-F、Bb2-AB-FsB-R和Bb2-AB-FsB-F、Bb2-AB-FsA-R进行PCR,得到片段Bb2-AB-F1和Bb2-AB-F2,通过同源重组连接两片段,得到质粒Bb2-AB。质粒Bb2-BC、Bb2-CD、Bb2-DE、Bb2-EF、Bb2-FG、Bb2-GH使用了相同的构建方法。
2.1.2 元件插入
以毕赤酵母GS115基因组为模板,利用引物对Fs1-pAOX1、pAOX1-Fs2进行PCR,得到两端带有BsaI酶切位点和接头序列1、2的片段pAOX1。之后配制以下体系:片段100 ng,Bb1-12 40 ng,Ligation mix 5 μL,BsaI 1 μL,加水补齐至10 μL。按照以下程序进行反应:37 ℃,3 h;50 ℃,5 min;80 ℃,10 min。之后转化大肠杆菌JM109,涂布氯霉素抗性平板筛选,挑取白色菌落并测序。之后用相同的方法构建了Bb1-pFLD1。Bb1-RPS25tt的构建方法相同,但使用的骨架质粒为Bb1-34。
代谢工程改造常需要筛选不同的外源基因。游离表达转化效率高、操作简单、对宿主影响小,先通过游离表达筛选出最适的外源基因再整合至基因组可以简化基因操作。因此构建了质粒Bb2-AB-CEN/ARS-BleoR以实现外源基因的游离表达。以质粒pYTK081为模板,利用引物对Fs1-CENARS-BleoR-F、CENARS-BleoR-R进行PCR。以pMCO为模板,利用引物对CENARS-BleoR-F、CENARS-BleoR-Fs4进行PCR。将以上获得的两个PCR产物进行融合PCR,得到片段CEN/ARS-BleoR,两端带有BbsI酶切位点和1、4接头序列。使用内切酶BbsI将其与Bb2-AB进行golden gate克隆,挑取白色菌落并测序。
毕赤酵母中抗性标记有限,对其进行多轮基因操作需要回收筛选标记。另一方面,出于安全性的考虑,生产所用的菌株不应带有抗性。LI等[11]构建了质粒pMCO,用于毕赤酵母中抗性标记的回收。以质粒pMCO为模板,利用引物对Fs1-Cre-F、BleoR-Fs 4进行PCR,得到片段的Cre-BleoR两端带有BbsI酶切位点和1、4接头序列,如图3所示。使用内切酶BbsI将其与Bb2-AB进行golden gate克隆,挑取白色菌落并测序,得到质粒Bb2-AB-Cre-BleoR。甲醇诱导Cre重组酶的表达后,loxP位点间的Cre和BleoR表达框被切除,即可消除抗性。
为实现实现目的基因的稳定表达,在Bb2-BC中插入了用于整合的元件AOX1TT,构建质粒Bb2-BC-AOX1TT。以毕赤酵母GS115基因组为模板,利用引物对Fs1-AOX1TT、AOX1TT-Fs4进行PCR,得到699的片段两端带有BbsI酶切位点和1、4接头序列。使用内切酶BbsI将其与Bb2-BC进行golden gate克隆,挑取白色菌落并测序。

图3 片段Cre-BleoR示意图Fig.3 Map of fragment Cre-BleoR
2.2 毕赤酵母多基因表达系统的应用
2.2.1 解除关键酶的负反馈抑制
芳香族氨基酸生物合成途径存在严格的调控机制,其中磷酸烯醇式丙酮酸(phosphoenolpyruvic acid,PEP)和4-磷酸赤藓糖(erythritose 4-phosphate,E4P)缩合生成2-酮-3-脱氧-D-阿拉伯庚酮-7-磷酸(2-ketone-3-deoxy-D-arabheptanone-7-phosphate,DAHP)及分支酸转变为预苯酸是2个关键的限速步骤,受到芳香族氨基酸的负反馈调节[13],通过定点突变或删除调节区域是解决这一问题的关键手段[14]。
2.2.1.1 游离型多基因表达载体的构建及验证
选择了大肠杆菌(Escherichiacoli)来源的DHAP合酶AroGD146Np(GenBank:WP_001109196.1)和分支酸变位酶PheAfbrp(GenBank:NP_311489.1)[15]及酿酒酵母(Saccharomycescerevisiae)来源的DHAP合酶Aro4K229Lp(Gene ID:852551)和分支酸变位酶Aro7G141Sp(Gene ID:856173)在毕赤酵母中进行游离表达。质粒构建流程如图4所示,以质粒Bb3-CEN/ARS-Aro4K229L-Aro7G141S的构建为例,以酿酒酵母BY4741基因组为模板,利用3对引物:Fs2-Aro4和Aro4-BsaI-R;Aro4-BsaI-F和Aro4K229L-rv;Aro4K229L-fw和Aro4-Fs3得到3段PCR产物,之后利用引物对Fs2-Aro4和Aro4-Fs3进行融合PCR。将所得PCR产物连接到质粒Bb1-23得到质粒Bb1-Aro4K229L,将其与Bb1-pAOX1、Bb1-RPS25Att、Bb2-BC进行golden gate组装,得到质粒Bb2-BC-Aro4K229L。用相同的方法得到质粒Bb2-CD-Aro7G141S,将其与Bb2-AB-CEN/ARS、Bb2-BC-Aro4K229L进行golden gate组装,转化大肠杆菌,挑取白色菌落,得到质粒Bb3-CEN/ARS-Aro4K229L-Aro7G141S,质粒图谱如图4所示。用相同的方法得到Bb3-CEN/ARS-AroGD146 N-PheAfbr。
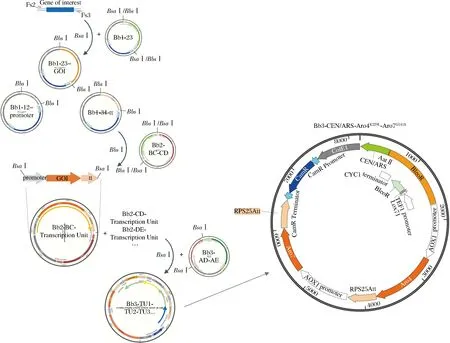
图4 游离型多基因表达载体的构建Fig.4 Construction of episomal multigene constructs
2.2.1.2 解除负反馈抑制对毕赤酵母合成2-PE的影响
分别将质粒Bb3-CEN/ARS-Aro4K229L-Aro7G141S和Bb3-CEN/ARS-AroGD146 N-PheAfbr电转至毕赤酵母GS115,得到菌株PE-1和PE-2。经过摇瓶发酵和产物测定,结果如图5所示,发现两种来源的酶都能有效解除毕赤酵母中的负反馈抑制,重组菌株产量都在150 mg/L以上,且酿酒酵母来源的ARO4K229L和ARO7G141S两基因效果更加显著。而出发菌株的产量始终在40 mg/L以下,低于线性范围(结果未展示)。以上结果证明过表达解除反馈抑制的关键酶可以有效解除终产物产生的反馈抑制。
2.2.2 过表达莽草酸途径
为了实现稳定表达,将酿酒酵母来源的基因ARO4K229L和ARO7G141S整合到毕赤酵母基因组中。为进一步促进毕赤酵母合成2-PE,同时将莽草酸途径中的DHAP合酶ARO3(Gene ID:8200288)、ARO5(Gene ID:8198396)和莽草酸途径下游的预苯酸脱水酶PHA2(Gene ID:8200719)基因整合到基因组中进行了过表达。
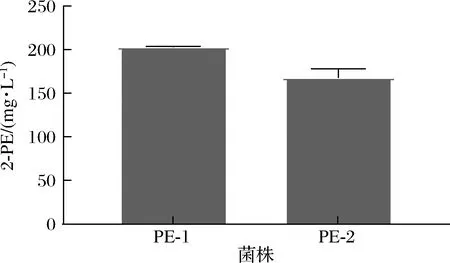
图5 菌株PE-1和PE-2的发酵结果Fig.5 Fermentation performance of strains PE-1 and PE-2
2.2.2.1 整合型多基因表达载体的构建及验证
使用2.2.1.1中描述的方法构建了质粒Bb2-CD-Aro4K229L、Bb2-DE-Aro7G141S、Bb2-EF-Pha2、Bb2-FG-Aro3、Bb2-GH-Aro5。将以上质粒与Bb3-AH、Bb2-AB-Cre和Bb2-BC-AOX1TT进行golden gate组装,将转化大肠杆菌后统计平板上绿色和白色菌落的个数。结果如图6-a所示,平板白色菌落占90%以上。菌落PCR验证结果如图,利用4对引物:(1)Fs2-Aro4、Aro7-Fs3;(2)Fs2-Aro7、Pha2-Fs3;(3)Fs2-Pha2、Aro3-Fs3;(4)Fs2-Aro3、Aro5-Fs3分别得到大小为2 756、2 543、2 876、3 374 bp的条带,如图6-b所示,证明质粒Bb3-Aro4K229L-Aro7G141S-Pha2-Aro3-Aro5构建成功,质粒图谱如图6-c所示,证明本研究所构建系统可完成多达7个元件的组装。
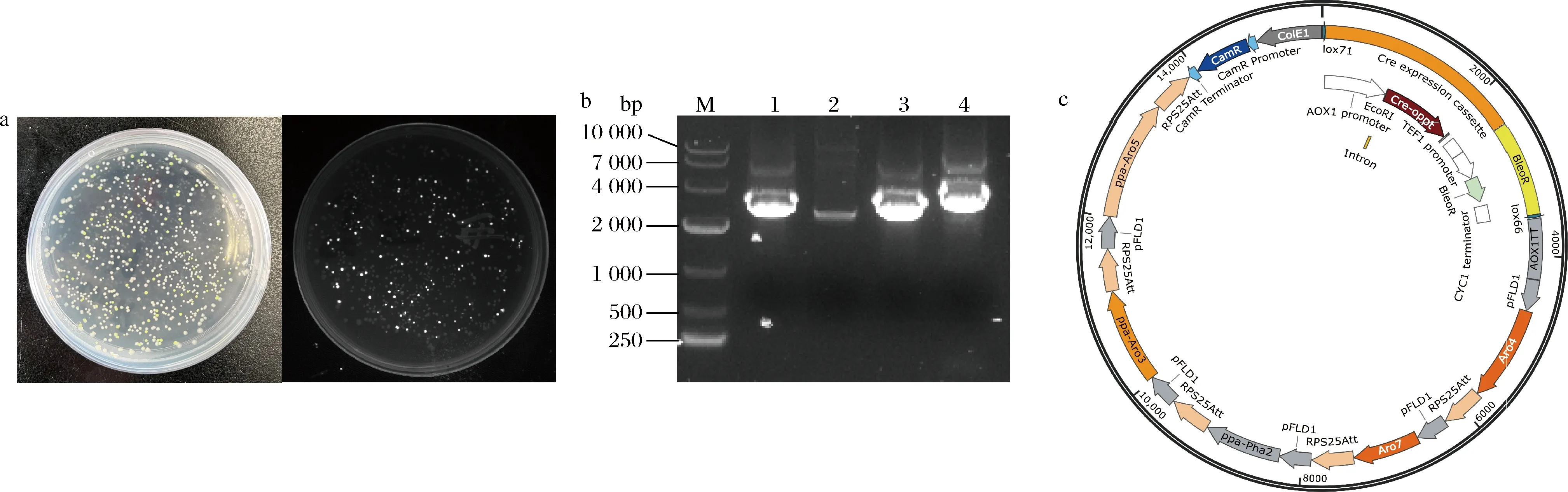
a-平板白色与绿色菌落分布;b-质粒Bb3-Aro4K229L-Aro7G141S-Pha2-Aro3-Aro5验证图(M-DL 10000 Marker;1-基因ARO4与ARO7连接验证; 2-基因ARO7与PHA2连接验证;3-基因PHA2与ARO3连接验证;4-基因ARO3与ARO5连接验证);c-质粒Bb3-Aro4K229L-Aro7G141S-Pha2-Aro3-Aro5图谱图6 游离型多基因表达载体的构建Fig.6 Construction of episomal multigene constructs
2.2.2.2 过表达莽草酸途径对毕赤酵母合成2-PE的影响
用XhoI对质粒Bb3-Aro4K229L-Aro7G141S-Pha2-Aro3-Aro5进行线性化,之后电转至毕赤酵母GS115,得到菌株PE-3。经过摇瓶发酵和产物测定,结果如图7所示,PE-3合成了408.4 mg/L的2-PE,是出发菌株的10倍以上。以上结果证明,引入Aro4K229Lp和Aro7G141Sp并高表达Pha2p、Aro3p、Aro5p是提高毕赤酵母2-PE合成能力的有效策略。
3 结论与讨论
基于golden gate组装的基因操作工具已在多种酵母中得到应用[16-18],本研究以此为基础,建立了一种毕赤酵母多基因组装系统,进行了7个原件的连接,转化后平板白色菌落在90%以上,说明该系统可以实现多片段的高效组装。使用该方法构建质粒可通过直观的荧光变化筛选到阳性克隆,菌株抗性可消除,目的基因的表达方式、启动子、终止子、整合位点均可灵活调整。利用MGAS,对毕赤酵母2-PE合成途径进行了一系列改造:通过游离表达不同来源的关键酶,解除负反馈抑制;整合表达ARO4K229L、ARO7G141S、PHA2、ARO3、ARO5,增加了莽草酸途径通量,最终构建的重组菌株2-PE产量达到了408.4 mg/L。本研究可为毕赤酵母代谢工程提供一种高效的多基因组装方法,也为毕赤酵母合成高附加值代谢物提供了参考。

a-PE-3发酵液中2-PE浓度;b-PE-3生长曲线图7 菌株PE-3的发酵结果Fig.7 Fermentation performance of strains PE-3
