流感病毒继发细菌感染加重的免疫学机制研究进展①
2023-01-19胡子琪张须龙首都医科大学基础医学院免疫学系北京100069
胡子琪 张须龙(首都医科大学基础医学院免疫学系,北京 100069)
流感病毒是造成全球季节性呼吸道疾病的重要病原体,是一种具有高度传染性的RNA病毒。虽然多数感染患者能自愈,但部分患者会出现急性肺损伤、急性呼吸窘迫综合征或继发细菌性肺炎而致死[1]。流感大流行和季节性流行期间,流感病毒继发细菌感染导致的细菌性肺炎是导致患者死亡的首要原因[2-4]。流感病毒感染可损伤呼吸道上皮细胞,易继发细菌感染,导致重症肺脏免疫损伤、呼吸窘迫,甚至死亡。流感病毒继发细菌感染急性加重性肺炎已得到广泛研究,其加重的机制,涉及宿主、病毒和细菌等多因素[5-6]。本文就流感病毒继发细菌感染的流行病学和易感机制的研究进展进行综述。
1 流行病学研究
据美国疾病控制与预防中心估计,在2017−2018年的流感季,美国共有4 880万人发生感染,7.94万人因流感死亡,是自2009年流感大流行以来的最高发病率和病死率,而全球估计每年有29.1~64.5万人死于流感相关的呼吸道感染[7-8]。历史上共有4次流感大流行,在1918年流感大流行期间,超过5 000万人死亡,而95%以上的重症和死亡是由于流感病毒继发细菌(主要是肺炎链球菌)感染[9]。1957−1958年大流行期间,即使存在抗生素,继发细菌性肺炎造成的死亡比例仍高达44%,且75%的死亡病例有金黄色葡萄球菌感染的细菌学证据[10]。1968−1969年大流行期间,肺炎的发病率与1957年相当,肺炎链球菌再次成为主要病原体[11]。2009年大流行期间,患者病死率与季节性流感相似,住院患者中约有一半出现细菌性肺炎,死亡病例以年轻人居多,且继发细菌感染的比例为25%~50%,肺炎链球菌和金黄色葡萄球菌感染是最常见的病因[12-15]。因此,流感病毒继发细菌感染是流感流行期间致死的重要原因。
2 易感机制
流感病毒和细菌共感染是否导致高病死率首先取决于两种病原体的感染顺序和时间[16]。在患者和小鼠的感染模型中,肺部流感病毒的滴度于感染后3~5 d达到峰值,第6天肺组织损伤最严重,在10~12 d病毒被清除。而继发细菌感染的最高易感时间从流感病毒感染后的第7天左右开始,持续约1周,说明此易感性是由于机体先天性免疫功能受损所引起,而非适应性免疫应答[17-19]。此外,由于宿主与病原体相互作用的复杂性,使免疫机制在联合感染的发展中起关键作用,其中抗病毒免疫虽然对病毒清除至关重要,但也可能增加继发细菌感染的易感性,如流感病毒感染诱导过度炎症反应损伤肺上皮细胞,从而破坏肺脏上皮屏障,增加细菌结合位点;流感病毒感染晚期肺脏固有免疫应答被抑制,导致抗菌水平降低,这些都被认为是促进细菌共感染的原因[17]。而最近的研究更多集中在流感病毒感染诱导的抗细菌先天免疫功能受损(图1),本文将对肺脏天然免疫细胞和细胞因子在流感病毒继发细菌感染中的作用及所诱导的细菌性肺炎的机制进行归纳和总结。
2.1 天然免疫细胞
2.1.1 巨噬细胞 肺脏巨噬细胞主要为驻留在肺泡内的肺泡巨噬细胞(alveolar macrophage,AM),还有少量驻留在肺实质的间质巨噬细胞(interstitial macrophage,IM)。而AM是肺脏首先接触流感病毒和细菌的免疫细胞,构成抵御细菌入侵的第一道防线[20-22]。
在天然免疫应答方面,一些研究认为流感病毒感染过程中肺脏AM数量基本不变,但诱导的调节因子抑制了AM的抗菌免疫能力,继而增加宿主对继发细菌感染的易感性。如SUN等[18]发现流感病毒感染小鼠T细胞产生的IFN-γ可通过下调AM的A类清道夫受体MARCO表达抑制其吞噬功能。而WU等[22]研究发现通过靶向Nrf2信号或Akt-TFEBMARCO途径可增加MARCO表达,可能是流感病毒继发细菌性肺炎的有效治疗方法。还有研究揭示流感病毒继发细菌感染后,STAT2信号通路通过调节C型凝集素受体(C-type lectin receptors,CLRs)和诱导型一氧化氮合酶(inducible nitric oxide syn‐thase,iNOS)表达,抑制巨噬细胞活化,导致机体对细菌的杀伤和清除不足[23](图1)。
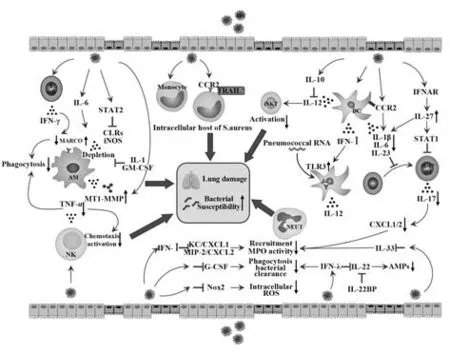
图1 天然免疫细胞和细胞因子在流感病毒继发细菌感染中的免疫机制Fig.1 Immunological mechanism of innate immune cells and cytokines in secondary bacterial infection following influenza virus
但也有一些研究证明流感病毒在一定时期内可通过诱导细胞凋亡介导AM耗竭,导致肺部清除细菌能力明显不足[21,24-25]。而BANSAL等[26]发现IL-1信号通路可阻止AM的耗竭,在宿主抵抗流感病毒和肺炎球菌共感染中起关键作用。此外,重组粒细胞-巨噬细胞集落刺激因子(GM-CSF)局部治疗或在肺中过表达GM-CSF能在细菌感染早期适度增加AM和中性粒细胞浸润,激活胞内活性氧(reactive oxygen species,ROS),增强其抗菌能力并改善小鼠生存率[27-28]。
另外在宿主耐受性损伤方面,膜Ⅰ型基质金属蛋白酶(membrane typeⅠmatrix metalloprotease,MT1-MMP)是流感病毒感染中显著升高的细胞外基质(extracellular matrix,ECM)重构胶原酶。病毒感染后肺部MT1-MMP主要来源于巨噬细胞,选择性抑制MT1-MMP可保护组织免受感染相关的组织损伤,维持基底膜的完整性,阻止继发细菌感染,预防败血症[29]。
综上所述,流感病毒感染通过耗竭AM,或损伤AM的抗菌功能以及促进其产生MT1-MMP损伤基底膜,降低早期检测和杀菌能力,增加宿主对细菌的易感性。
2.多音型过腔。如“昆南”中阴平声字“惊”的唱调(《牡丹亭·游园》【醉扶归】“沉鱼落雁鸟惊喧”,706)。其中的过腔即为多音。“昆北”中阳去声字“弄”的唱调(《长生殿·絮阁》【喜迁莺】“弄鬼装妖”,777),其中的过腔即为多音型过腔。
2.1.2 单核细胞 外周血中单核细胞至少存在两个主要亚群,一种是经典的Ly6Chi单核细胞,也称炎症单核细胞、渗出性巨噬细胞(exudate macrophage,exMAC)、产生TNF-α/iNOS的树突状细胞(TNF-α/iNOS-producing DC,TipDC)或TRAIL+单核细胞,其特征是趋化因子受体2(CCR2)高表达,并在流感病毒继发细菌感染中起重要作用[30-32]。另一种是非经典的Ly6Clo单核细胞,为抗炎单核细胞,缺乏CCR2的表达,小鼠外周血中还含有Ly6Cint单核细胞,为单核细胞分化的中间产物[33-34]。
流感病毒感染可招募CCR2+炎症单核细胞到肺脏,增强肺部炎症、清除病毒,但也同时加重肺脏免疫损伤,敲除CCR2可显著提高流感病毒感染后的生存率[30]。而在后续细菌感染加重机制中,ELLIS等[32]发现这些炎症单核细胞通过一种依赖于肿瘤坏死因子相关凋亡诱导配体(TNF-related apoptosisinducing ligand,TRAIL)的机制,在流感病毒感染后损伤肺部屏障,有助于增加细菌定植,促使细菌侵袭全身。此外,还有研究发现流感病毒募集的炎症单核细胞可作为金黄色葡萄球菌胞内存活的宿主,从而提高了金黄色葡萄球菌对抗生素的耐药性[35]。以上结果表明炎症单核细胞由于对病毒和细菌清除的相互冲突导致了联合感染加重。
2.1.3 中性粒细胞 中性粒细胞是宿主清除细菌的关键细胞,而抑制中性粒细胞募集是流感病毒感染后易继发细菌感染的关键致病机制,流感病毒诱导的Ⅰ型IFN(IFN-Ⅰ)及下游信号,可通过抑制IL-17介导免疫应答抑制中性粒细胞的募集和功能[36-37]。在继发肺炎链球菌中,IFN-Ⅰ选择性地抑制中性粒细胞趋化因子KC/CXCL1和MIP-2/CXCL2的产生,导致抗细菌感染的早期阶段中性粒细胞应答低下[19]。流感病毒还可抑制粒细胞集落刺激因子(GCSF)分泌、降低髓过氧化物酶(myeloperoxidase,MPO)活性,造成中性粒细胞吞噬和清除细菌的功能障碍,引发细菌感染[38]。
中性粒细胞可能是肺脏金黄色葡萄球菌感染最重要的吞噬细胞亚群[39-40]。中性粒细胞内NADPH氧化酶介导吞噬和杀菌过程中ROS的产生,称为氧化爆发,而中性粒细胞具有比巨噬细胞更强的氧化爆发能力。NADPH氧化酶主要的功能亚基NADPH氧化酶2(nicotinamide adenine dinucle‐otide phosphate oxidase 2,Nox2)在吞噬细胞中的活性对清除金黄色葡萄球菌至关重要,而流感病毒感染抑制NADPH活性,减少吞噬细胞内ROS水平,降低其细菌清除能力[41]。后续研究也发现Nox2不但介导抗菌反应,而且也参与组织炎症性损伤。在合并感染时,Nox2诱导的氧化应激增加炎症细胞的坏死死亡,对周围组织造成致命损伤,联合使用抗生素和NADPH氧化酶抑制剂可显著提高存活率[42]。因此,Nox2的激活在共感染时具有双刃剑作用,流感病毒感染破坏依赖于Nox2的抗菌免疫和炎症之间的平衡,既增加细菌感染的易感性,又导致广泛的肺损伤。
2.1.4 树突状细胞(dendritic cell,DC)DC是促炎细胞因子IL-12的主要来源,诱导Th1细胞分化并激活其他天然免疫细胞产生细胞因子,在调节天然和适应性免疫反应方面发挥核心作用。单核细胞来源的DC(monocyte-derived DC,moDC)是感染肺脏最常见的DC,可激活并诱导初始T细胞增殖分化,在宿主防御细菌性肺炎中起重要作用[30]。流感病毒感染的人moDC分泌IFN-Ⅰ和其他可溶性因子,继而显著上调周围DC的TLR3表达,通过识别结合肺炎链球菌RNA增强IL-12分泌,是继发肺炎链球菌感染诱导DC分泌IL-12的关键机制之一[43]。
与单核细胞相似,GURCZYNSKI等[44]发现流感病毒感染的ccr2-/-DC产生高水平的IL-6和IL-23,诱导Th17分化和IL-17产生,以及肺泡上皮细胞IL-17R上调,从而促进CXCL1/2产生,趋化更多中性粒细胞,靶向CCR2可能是调控流感病毒继发细菌感染易感性的潜在治疗策略。
2.1.5 自然杀伤(natural killer,NK)细胞NK细胞既能清除病毒感染细胞,又能分泌抗病毒细胞因子,是抑制流感病毒感染的重要细胞[45]。SMALL等[46]研究发现,流感病毒感染抑制NK细胞分泌TNF-α的水平,并抑制继发细菌感染中NK细胞的募集和活化信号,继而抑制AM对金黄色葡萄球菌的吞噬作用。说明肺脏NK细胞分泌TNF-α的功能减弱不但降低自身杀伤功能,而且抑制AM的抗菌活性,是流感病毒继发细菌感染的关键机制之一。
2.2 细胞因子 过度免疫应答是重症流感病毒感染高病死率的关键机制,而细胞因子是炎症反应的关键调节因子,并在机体流感病毒继发细菌性肺炎的发生发展中发挥重要作用,研究发现流感病毒诱导的多种细胞因子对继发细菌性肺炎易感性有重要影响[47]。
流感病毒诱导的IFN-Ⅰ可通过抑制Th17型免疫、降低细菌清除能力而增加继发感染易感性[1]。STAT1是IFN下游信号转导中的关键转录因子,介导IAV感染诱导的宿主炎症反应[48]。在流感病毒继发MRSA感染中,LEE等[37]研究发现,stat1-/-小鼠的Th17型免疫极化增加,证明STAT1信号通路通过调控Th17型免疫极化,调节呼吸道细菌感染的关键细胞因子家族Th17相关细胞因子的产生,介导流感病毒继发细菌感染的易感性。
流感病毒感染诱导的IFN-γ激活单核细胞、NK细胞和巨噬细胞,增强组织炎症反应,但抑制中性粒细胞募集和活化,降低肺脏清除胞外细菌感染的能力[18,48]。与IFN-Ⅰ相似,流感病毒诱导的IL-13和IFN-γ之间也存在感染不同时间影响继发细菌感染易感性,在流感病毒感染后3 d内,IL-13通过抑制IFN-γ而增强细菌清除能力;而在病毒感染第7天,IL-13诱饵受体IL-13Rα2水平的增加抑制了IL-13信号传导,进而促进IFN-γ信号,在一定程度上加重细菌性肺炎[49]。
Ⅲ型IFN(也称IFN-λ、IL-28和IL-29)是呼吸道上皮细胞最早产生的主要IFN[50-51]。高表达的Ⅲ型IFN可导致上呼吸道微生物群的增加和重组,而敲除IFN-λ受体降低小鼠在MRSA单独或联合感染的细菌载量,其机制为增加IL-22及中性粒细胞明胶酶相关脂质运载蛋白(neutrophil gelatinase-associated lipocalin,NGAL)等抗菌肽(antimicrobial peptides,AMPs)表达[52]。IFN-λ还可通过抑制中性粒细胞的迁移和吞噬功能减少对细菌的摄取[53]。
2.2.2 白介素(interleukin,IL)
2.2.2.1 IL-1 IL-1在重症流感病毒感染时加重急性肺脏免疫病理反应,但在抵御继发性肺炎链球菌感染中起保护作用。在共感染小鼠模型中,IL-1增强肺脏免疫细胞对细菌的清除能力,敲除IL1R1显著增加细菌载量和死亡率[26]。
2.2.2.2 IL-6流感病毒感染以及继发细菌感染后,IL-6的水平显著升高,激活多种免疫细胞介导的炎症反应[16,54]。继发细菌感染后,IL-6诱导巨噬细胞表面MARCO表达增加,增强其吞噬能力,降低继发肺炎链球菌感染的易感性,而IL-6敲除小鼠肺脏细菌载量增加,加重肺损伤并伴有高死亡率[55]。
2.2.2.3 IL-10 IL-10是流感病毒感染过程中产生的介导免疫抑制的重要抗炎因子,流感病毒感染后产生的过量IL-10抑制中性粒细胞的功能而易引发呼吸道肺炎链球菌感染,流感病毒诱导的肺脏IL-10也抑制moDC产生IL-12,抑制恒定自然杀伤T(invariant natural killer T,iNKT)细胞的激活,降低其抗细菌感染能力[56-57]。因此IL-10是流感病毒感染后促进细菌性肺炎发生的重要细胞因子。
2.2.2.4 IL-22流感病毒感染后诱导肺脏产生IL-22及相关细胞因子。在继发细菌感染中,IL-22可通过增强呼吸道上皮完整性和诱导AMPs产生,促进肺脏抵御继发细菌感染[58]。可溶性诱饵受体IL-22结合蛋白(IL-22-binding protein,IL-22BP)抑制IL-22功能或敲除IL-22的小鼠流感病毒感染模型中,因气道上皮完整性降低而损害上皮屏障功能,加重继发细菌感染诱导的肺损伤[59]。
2.2.2.5 IL-27流感病毒感染后通过IFNAR信号通路诱导IL-27产生。IL-27不但下调肺炎链球菌激活的DC分泌IL-23和IL-1β,而且通过STAT1调控γδT细胞活性、抑制其IL-17A的产生,抑制中性粒细胞的募集和其胞内MPO的活性,降低对继发肺炎链球菌的固有免疫应答[36]。因此流感病毒诱导的IL-27信号促进了继发细菌性肺炎的发展。
2.2.2.6 IL-33 IL-33具有促炎和抗炎的双重作用。在感染和过敏性疾病模型中,IL-33通过诱导IL-5和IL-13等促进Ⅱ型免疫应答[60-61]。而在流感病毒感染模型中,IL-33的分泌被抑制,减少了中性粒细胞向肺脏黏膜屏障募集,降低肺脏对继发细菌感染的清除能力,因此IL-33在机体抗菌黏膜屏障免疫中起关键作用[40]。
3 总结与展望
肺脏固有免疫系统可通过协调多种免疫细胞和细胞因子的反应,有效抵御肺脏细菌感染而维持免疫平衡。而先前的流感病毒通过多种机制影响AM的数量与功能,募集炎症单核细胞,抑制中性粒细胞和NK细胞的趋化与活化,以及影响多种细胞因子间的相互作用,降低机体肺脏抗菌免疫应答,损伤肺部屏障,利于继发细菌的入侵和定植,导致严重的肺脏损伤。
目前临床上鲜有治疗流感继发细菌感染的方法,主要是联合抗流感药物和多种抗生素用药。但由于细菌多重耐药性的逐渐增加,抗生素的效果越来越差,而接种细菌疫苗也不能很好地降低流感后细菌性肺炎的发生[53,62]。不过目前有研究发现一种依赖IFN的调节机制,在保护抗菌功能的同时防止过度免疫病理反应,而当重新激活肺脏天然免疫应答可显著降低继发细菌感染的易感性,可能是一种新的预防和治疗流感病毒继发细菌感染的免疫调节策略[48,28]。此外,如前所述无论是IFN-Ⅰ信号表达的差异,还是IL-13对IFN-γ的作用,在流感病毒感染的不同时间具有不同的作用,合适的干预时间和靶点是流感病毒继发细菌感染诱发细菌肺炎免疫治疗的关键因素,错误的干预时间反而可能增加细菌的易感性。另外,免疫细胞及相关分子在病毒感染和细菌感染中的作用也不同。因此,得当的治疗时间和合适的治疗靶点是流感病毒继发细菌感染免疫治疗的重要因素。
综上所述,深入了解肺脏天然免疫细胞和细胞因子在流感病毒继发细菌感染中的作用及机制,有助于发现新的病毒继发细菌感染治疗靶点、开发潜在的免疫疗法治疗途径、降低病毒继发细菌感染的发病率、提高病毒继发细菌感染患者的存活率。
