稳定同位素稀释-增强脂质去除净化-联用 气质法快速测定水产品中的多环芳烃
2023-01-15何雯倩马承鸿叶日金
何雯倩,韦 誉,马承鸿,叶日金
(1.广东省食品工业公共实验室,广东广州 511442;2.广东省食品工业研究所有限公司,广东广州 511442; 3.广东省质量监督食品检验站,广东广州 511442)
多环芳烃(Polycyclic Aromatic Hydrocarbons,PAHs) 是煤、石油、木材、烟草和有机高分子化合物等有机物不完全燃烧时产生的挥发性碳氢化合物,是重要的环境和食品污染物[1-3]。由于多环芳烃的结构稳定,在环境中降解速度较慢[4-5],因此容易富集并通过食物链进入食品生产原料当中[6-7]。菲在多环芳烃中,由于三个环的中心不在同一直线上,因此对称性没有萘或蒽等化合物强,芳香性也更弱,同时由于其毒性较大,2017年被世界卫生组织国际癌症研究机构列为3类致癌物。报道称,多环芳烃具有很强的致癌、致畸、致突变等毒副作用[8-10],对组织器官有严重的损害和影响。因此,对食品原料或终端产品的质量安全问题进行有效监管具有重要的意义,而开发快速高效的分析方法是进行市场监管的有效手段。
目前,多环芳烃的检测方法主要有荧光法、气相色谱-质谱法[11-12]、气相色谱-串联质谱法[13-16]和高效液相色谱-荧光/紫外检测法[17-18]等。荧光法虽然选择性强, 但对同类化合物定性还需要出峰时间确证,尤其对于同分异构体,难以区分定性定量。高效液相色谱对多种化合物的分离时间长、分离难度大、方法定性需要核对每个化合物的出峰时间,步骤烦琐,且无法使用内标物对结果进行校正。质谱法具有高通量、分析时间短、定性定量准确的特点,因此被检测实验室广泛应用。本文使用增强脂质去除净化管(Enhanced Matrix Removal,EMR)对提取液进行净化,同位素内标进行定量校正,建立了快速测定水产品中多环芳烃菲的分析方法。该方法定量准确、回收率高、重现性好,为水产品中的多环芳烃检测研究提供技术参考。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器
7890B-5975C安捷伦气相色谱-质谱仪,配有电子轰击离子源(Electron impact ion source, EI);KQ-800DB超声波清洗仪,昆山超声仪器有限公司;Promax 2020摇床,德国海道夫公司;3-18k高速离心机,德国西格玛公司;Heidolph Multi Reax涡旋振荡器,德国海道夫公司;Hei-VAP Core ML/G3旋转蒸发仪,德国海道夫公司;MilliQ超纯水机,德国默克密理博公司。
1.1.2 试剂
甲醇、乙酸乙酯、乙腈,色谱纯,美国赛默飞世尔公司;无水硫酸镁,分析纯,广州化学试剂厂;C18、PSA固相萃取剂,分析纯,上海安谱实验科技股份有限公司;商品化增强脂质去除净化管(Enhanced Matrix Removal,EMR),分析纯,安捷伦科技有限公司;多环芳烃菲和菲-d10标准溶液,100 mg·L-1,天津阿尔塔科技有限公司。
1.2 标准溶液的配制
①外标溶液。取1 mL菲标准溶液,正己烷溶解并定容至100 mL容量瓶,配制成1 mg·L-1的标准储备溶液。②内标储备溶液。取1 mL菲-d10标准溶液,正己烷溶解并定容至100 mL容量瓶,配制成1 mg·L-1的标准储备溶液。③标准工作曲线。取上述菲标准储备溶液使用正己烷配制5.0 μg·L-1、10.0 μg·L-1、20.0 μg·L-1、50.0 μg·L-1、100.0 μg·L-1和200.0 μg·L-1的校准曲线(各点含内标浓度为20.0 μg·L-1)。
1.3 前处理方法
①取样。取约200 g水产品可食用部分均质, -18 ℃冷冻保存。②提取。称取5 g均质后的样品于50 mL离心管中,加入20 μL浓度为1 mg·L-1的内标溶液,加入15 mL乙腈,超声提取15 min,以 8000 r·min-1离心5 min,转移上清液至梨形瓶中。重复提取1次,合并提取液于梨形瓶中。40 ℃旋转蒸发至2~3 mL,待净化。③净化。EMR管用 2 mL水活化,将待净化液转移至管中,梨形瓶使用2 mL乙腈洗涤一次,并将洗涤液转移至净化管中,涡旋振荡2 min,以4000 r·min-1离心5 min。净化液用2 g无水硫酸镁于15 mL离心管中除水后, 4000 r·min-1离心5 min,上清液转移至氮吹管中,40 ℃氮吹至近干,用1 mL正己烷复溶,过0.22 μm有机相滤膜后,供气相色谱-质谱仪测定。
1.4 色谱及质谱条件
1.4.1 色谱条件
安捷伦DB-5ms石英毛细管柱(30 m×0.25 mm,0.25 µm);升温程序:80 ℃保持1 min,10 ℃·min-1升温至180 ℃保持1 min,20 ℃·min-1升温至280 ℃保持7 min;载气流速:1.3 mL·min-1;分流进样:分流比2∶1;接口温度:300 ℃;进样口温度:290 ℃;溶剂延迟:8 min;进样量:1 µL。
1.4.2 质谱条件
电子轰击离子源;电子能量:70 eV;四极杆温度:150 ℃;离子源温度:230 ℃;溶剂延迟:8 min;选择离子采集模式(SIM);定量离子:m/z178,定性离子:m/z176、m/z152;内标定量离子:m/z188。菲和菲-d10内标的定量离子流色谱图见图1。
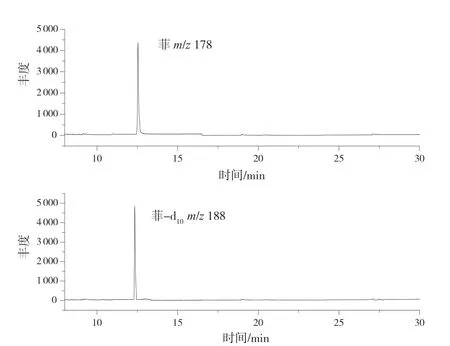
图1 菲和菲-d10内标定量离子流图
2 结果与分析
2.1 提取溶剂的选择
实验以均质好的阴性九节虾样品作为试样,标准溶液添加梯度为10 μg·kg-1,分别考察了甲醇、乙腈和乙酸乙酯的提取效率。结果显示,乙酸乙酯回收率较低,可能是因为乙酸乙酯极性较低,容易提取到脂溶性的杂质,对结果造成影响;甲醇和乙腈的回收率较好,但相对于甲醇,乙腈的回收率更突出,可能是由于菲的疏水性较强,更易溶于乙腈。实验最终选择乙腈作为提取液,回收率结果如图2 所示。
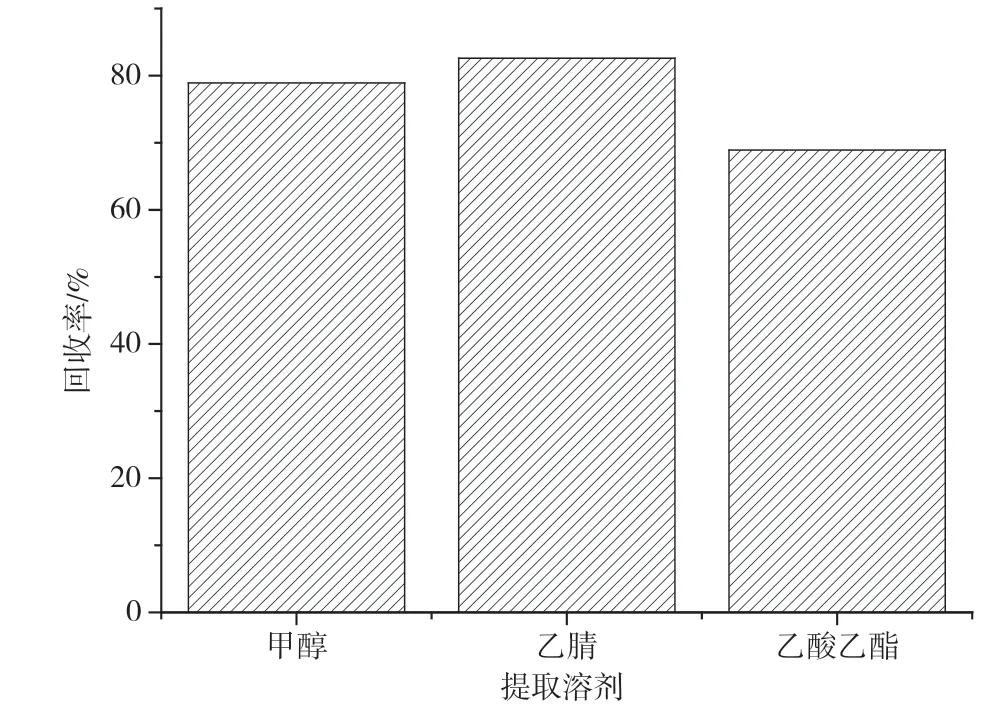
图2 不同提取溶剂的回收率结果
2.2 提取方式的选择
实验同时对比摇床,超声和涡旋等3种提取方式。以阴性九节虾样品作为试样,标准溶液添加梯度为10 μg·kg-1,使用乙腈进行提取。结果表明,摇床提取效率最低,回收率只有75.8%;其次是涡旋振荡的回收率为79.5%;回收率最好的是超声方式,回收率达到84.3%。其主要原因可能是因为普通的振摇模式,样品难以与提取溶剂充分接触,所以回收率较低,而超声通过高频率的振动,可使样品充分提取,因此最终选择超声方式进行提取,结果见图3。
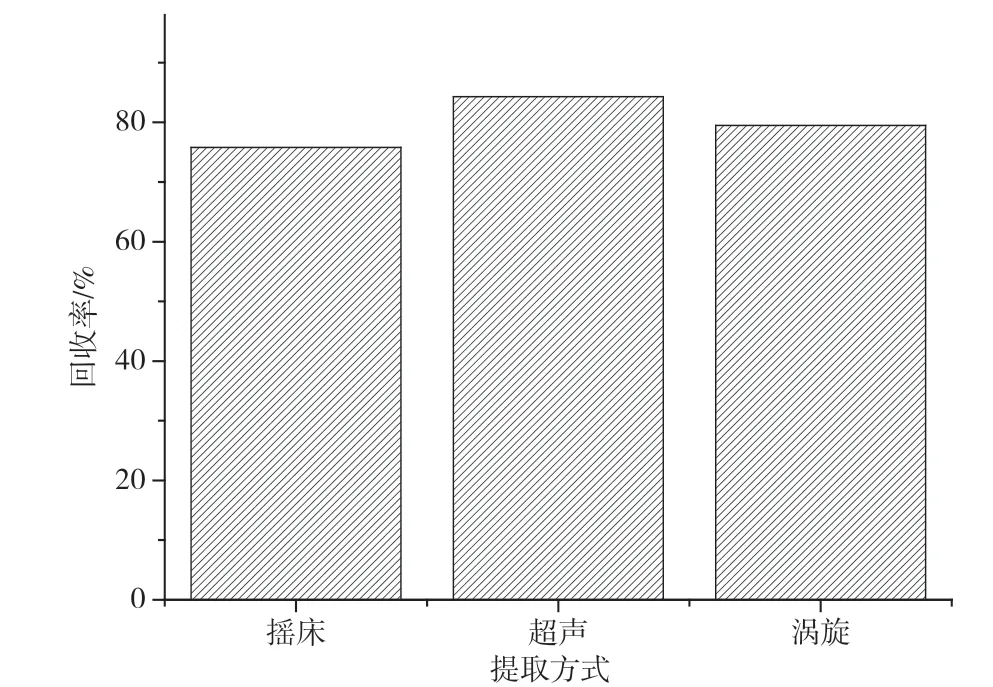
图3 不同提取方式的回收率结果
2.3 提取次数的确定
由于菲的脂溶性较强,而水产品中含有较多的脂肪,提取次数过少,可能造成提取不完全,影响回收率;若提取次数过多,则溶剂消耗量大,后续旋转蒸发处理耗时长,同时可能导致提取到样品中过多的油脂等低极性杂质,增大基质效应而影响回收率。实验分别对加标样品进行1次、2次和3次提取,结果发现,提取1次时,提取并不充分,回收率只有53.7%,而提取2次和3次的回收率则分别达到84.1%和84.5%。综合考虑,提取3次相比提取2次,回收率只提升0.4个百分点,而时间和试剂消耗却高出1.5倍,因此最终选择重复提取2次。
2.4 净化剂类型的选择
为了保护设备和降低基质效应,需要对提取液进行净化。水产品中的杂质多以蛋白质、氨基酸、脂肪以及一些血液和组织中的神经递质和其他内源物质为主。实验对比了EMR增强脂质去除净化管和固相含量同样为400 mg的C18和PSA,以阴性九节虾样品作为试样考察其回收率,标准溶液添加梯度为10 μg·kg-1。由图4可知,EMR增强脂质去除净化管的回收率最高,而C18和PSA的回收率相当。根据固相基团进行分析,可能是因为EMR可针对脂质进行吸附,较大程度上降低了干扰。C18和PSA虽然一定程度上也能对氨基酸和脂质等杂质进行吸附,但其选择性较低,导致回收率偏低。实验最终选用EMR增强脂质去除净化管净化。
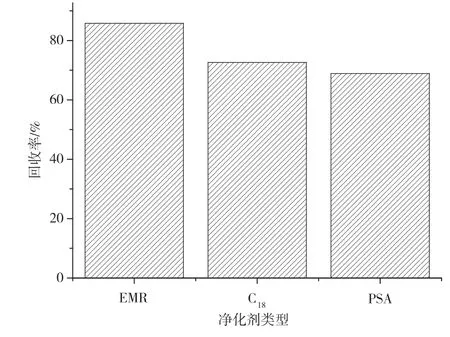
图4 不同净化剂类型的回收率结果
2.5 基质效应的讨论
基质效应分为基质增强效应和基质减弱效应两种类型,即基质对目标物质的检测信号有增强或抑制的作用,从而影响目标物质的最终定量结果。基质效应可根据公式(基质曲线斜率/溶剂曲线斜率-1)×100进行计算。实验以阴性九节虾样品作为空白基质,用其基质提取液和正己烷分别配制菲的外标物曲线,测定后经计算发现,基质效应为40,(绝对值小于20为弱基质效应,在20~50为中等基质效应,超出50为强基质效应),说明基质效应严重。内标法是抵消基质效应最有效的方法之一,因此实验最终使用菲的同位素内标菲d-10对结果进行 校正。
2.6 线性方程及灵敏度
由于前处理净化后,基质效应对结果的影响仍然明显,因此使用内标法进行定量,配制浓度为5.0~200.0 μg·L-1的标准曲线,其中内标浓度为20.0 μg·L-1,在实际的测定过程中,可根据样品的实际含量进行稀释或者浓缩,使测定值在曲线范围内。实验所测得的方程及相关系数见表1,同时以低浓度加标测定方法检出限和方法定量限,由表1可知,相关系数为0.9996,说明线性关系良好,曲线定量准确,检出限为0.3 μg·kg-1,定量限为1.0 μg·kg-1,方法整体灵敏度较高,达到测定要求。
2.7 精密度、回收率及重复性
选择阴性九节虾样品和草鱼样品进行加标回收率试验,以验证方法的回收率和稳定性,对样品进行1.0 μg·kg-1、2.0 μg·kg-1和10.0 μg·kg-1水平的加标,平行测定6次样品,结果如表2所示,九节虾和草鱼所测得回收率差异不大,回收率为78.7%~85.4%,相对标准偏差为2.52%~3.36%。表明该方法的回收率较高,且偏差小,详见表2。
2.8 方法实际测定应用
选择市场或者超市售卖的水产样品进行测定,最终检测了10个鱼样品,均未检出;10个虾样 品,1个检出3.55 μg·kg-1;10个螃蟹样品,1个检出 9.92 μg·kg-1。说明目前市面上的水产品受到了一定的污染,存在影响消费者健康安全的风险,应进一步加强监管力度,建立有效的管理手段。

表1 菲的线性方程和相关系数
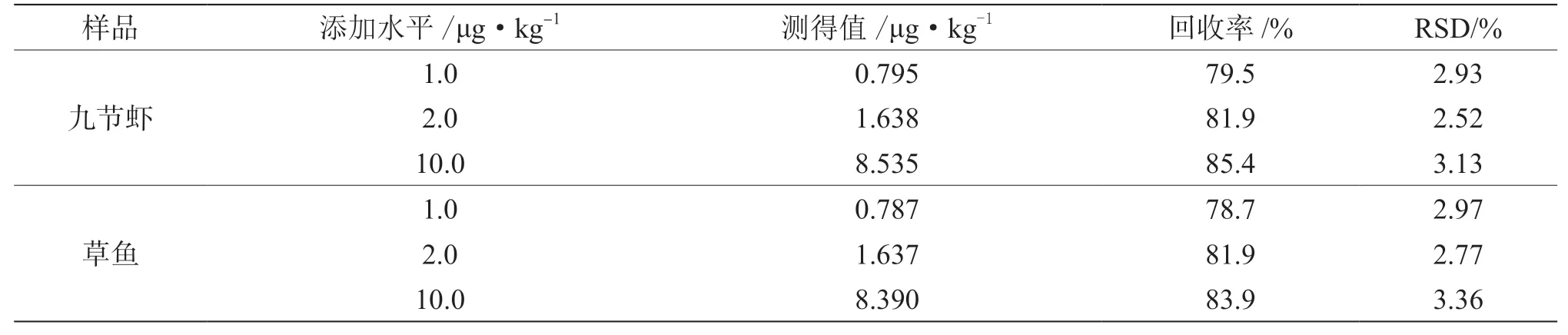
表2 回收率和相对标准偏差
3 结论
本文以菲作为探针化合物,建立了EMR增强脂质去除净化管结合气相色谱-质谱法测定水产品中多环芳烃残留量的分析方法。样品使用乙腈进行提取,经净化后,基质效应降低,且回收率较高,同时使用内标物校正结果,准确度好;目标物在短时间内完成出峰,峰形对称,柱效高;该方法检出限低,灵敏度高,回收率及相对标准偏差符合一般测定要求,对于不同样品,回收率均达到分析条件,可对水产品中多环芳烃的分析方法开发提供技术支持。
