E3泛素连接酶和小泛素蛋白样修饰蛋白在子痫前期发病机制中的研究进展
2023-01-13周天凡姚莉萍顾盛奕花晓琳
周天凡, 姚莉萍, 顾盛奕, 花晓琳
(1. 同济大学附属第一妇婴保健院产科,上海 201204; 2. 同济大学附属第一妇婴保健院超声科,上海 201204)
子痫前期(pre-eclampsia, PE)是妊娠中晚期出现的、一种以广泛血管内皮损伤为特点的妊娠期特有疾病。多项研究表明其病因和发病机制可能与各种高危因素引起的子宫螺旋小动脉重塑不足有关[1],导致滋养细胞和蜕膜病变引起胎盘低灌注,使得胎盘因子进入母体循环,从而引起内皮损伤以及系统性炎症反应的激活,最终导致PE。

1 泛素化修饰与PE
1.1 泛素化修饰概述
蛋白质泛素化修饰是指一个或多个泛素分子在一系列酶的作用下与底物蛋白质共价结合的修饰过程。一般泛素化修饰的底物蛋白会被泛素-蛋白酶体系统(ubiquitin-proteasome system, UPS)识别并降解,是下调蛋白表达的主要机制。UPS参与体内多种生理过程,包括转录调控、细胞周期、细胞凋亡、DNA损伤修复及代谢、免疫和炎症等。在人类细胞中有超过1 000种蛋白质调节泛素化[3],泛素化失调能够导致多种疾病,例如肿瘤、神经退行性疾病、肝脏疾病等。
整个泛素化过程可概括如下[4]: (1) E1泛素激活酶(E1)激活ATP和泛素;(2) E1将激活的泛素传递给E2泛素结合酶(E2),形成E2-泛素中间产物;(3) E3泛素连接酶(E3)同时结合E2与底物蛋白,并促使E2泛素分子转移到底物蛋白上[5];(4) 上 述过程不断重复,直到蛋白质上连接的多个泛素形成一条泛素链;(5) 真核生物体内负责降解蛋白质的26 s蛋白酶体识别被泛素化的底物蛋白并将其降解;(6) 去泛素化酶(deubiquitinase, DUBs)可逆地将蛋白从泛素分子上分离,避免蛋白过度降解并重新释放出泛素单体,使泛素重新进入泛素蛋白酶体的循环,从而调节生物体内蛋白的平衡,见图1。
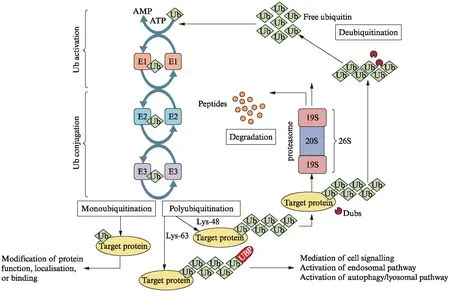
图1 泛素-蛋白酶体系统[6]Fig.1 Ubiquitin-proteasome system[6]
在泛素化修饰过程中,E3泛素连接酶通过特异性地识别并结合底物蛋白发挥了最为关键的作用。目前已知人类基因组中有多达600多种的特异性E3泛素连接酶。E3根据其生化和结构特点主要分为两大家族: RING结构域家族和HECT结构域家族。RING家族的E3泛素连接酶可以促进泛素从E2直接转移到底物蛋白,而HECT家族的E3泛素连接酶与同源的E2相互作用,然后与泛素形成硫酸盐连接,随后将泛素转移至底物蛋白[7]。本文将着重探讨E3泛素连接酶在PE发病机制中的作用。下面将阐述参与PE发病机制泛素化修饰的E3泛素连接酶。
1.1.2 SCFFBW2介导GCM1泛素化降解 SCF(skp1-cullin1-F-box)复合体属于RING家族的E3泛素连接酶,主要由SKP1(S-phase kinase-associated protein 1)、CUL1(cullin-1)、F-box蛋白组成。SCF复合体中由F-box蛋白特异性识别底物并针对底物进行泛素化,随后调节细胞周期、细胞增殖、细胞凋亡、血管生成和转移等过程。F-box蛋白有3个主要的亚家族: FBXW、FBXL和FBXO亚家族[8]。
FBW2(F-box and WD repeat domain containing 2 protein)属于FBXW家族。Yung等[9]研究发现,FBW2介导的泛素化促进胶质细胞缺失因子1(Glial cells missing homolog 1, GCM1)降解。GCM1是胎盘发育中重要的转录因子,通过调节胎盘生长因子、人合胞素1以及HtrA丝氨酸肽酶4的表达,在介导滋养层细胞分化中起关键作用。Chiang等[10]研究显示,低氧通过抑制磷脂酰肌醇3激酶-(phosphoinositide 3-kinases, PI3K)-蛋白激酶B(proteinkinase B, AKT)信号通路,激活下游的糖原合成酶激酶3β(glycogen synthase kinase 3 beta, GSK-3β),这是参与胎盘植入过程的重要信号通路之一[11]。被激活的GSK-3β对GCM1蛋白322位点的丝氨酸磷酸化修饰,进而招募FBW2,介导GCM1泛素化降解。因此在低氧情况下,胎盘中GCM1及其靶基因胎盘生长因子、人合胞素1的表达减少,而HtrA丝氨酸肽酶4表达增加,影响滋养细胞融合和胎盘血管生成,导致胎盘发育异常从而引起PE[12]。而GSK-3β的抑制剂LiCl可减少缺氧诱导的SCFFBW2介导的GCM1 泛素化降解,提示靶向PI3K-AKT-GSK-3β对于治疗PE潜在作用的相关机制。
1.1.3 SCFβ-TrCP介导Snail泛素化降解 β-转录重复包含蛋白(beta-transducin repeat-containing pro-teins, β-TrCP)也称为FWD1,属于FBXW蛋白家族。通过调节底物蛋白的降解,β-TrCP参与各种细胞过程,包括细胞迁移和侵袭。
上皮间质转化(Epithelial-Mesenchymal Transi-tion, EMT)是绒毛外滋养细胞获得迁移和浸润能力的重要过程,而转录因子Snail通过抑制E-钙黏蛋白(E-cadherin)的表达来促进EMT,与胚胎发育和肿瘤侵袭密切相关[13]。研究发现β-TrCP表达在PE中增加,介导其下游底物Snail发生泛素化降解,抑制滋养细胞的EMT过程使其浸润不足,导致胎盘浅着床并促进PE的发展。
另外,Wu等[14]的研究显示,β-TrCP过表达还可能通过泛素化降解NF-κB的抑制蛋白IκB以及β-连环蛋白来抑制血管内皮细胞生长因子受体2的表达。因此,β-TrCP表达升高不仅能抑制滋养细胞EMT,也可能导致血管生成相关因子缺乏,从而促进PE的发生发展。Wu等[15]的研究显示,miR-135a-5p通过靶向β-TrCP促进滋养层细胞的迁移和侵袭,提示靶向β-TrCP对于治疗PE的潜在机制。
细胞周期蛋白G2(cyclin G2 protein, CCNG2)通过诱导G1/S期停滞对细胞周期进程有明显的负性调控作用,过去有很多研究显示CCNG2可能与恶性肿瘤和糖尿病肾病等疾病有关。而近年来的研究显示,在PE胎盘中,CCNG2的表达增加并且与滋养细胞功能障碍有关。Sun等[16]的研究表明,PE中CCNG2过表达使得RNF123的水平增加,并且加强了RNF123与蓬乱蛋白2(dishevelled segment polarity protein 2, Dvl2)的结合,通过UPS诱导Dvl2过度降解。Dvl2是Wnt信号通路的转导枢纽,而Wnt信号通路对于调节凋亡、侵袭及氧化应激反应起着重要作用。因此Dvl2表达减少抑制了非经典Wnt/PCP-JNK信号通路及其下游效应物,包括EMT相关蛋白,因而导致滋养细胞浸润不足促进PE的发展。CCNG2/RNF123/Dvl2/JNK轴可能通过滋养细胞功能调节参与PE的发病机制和进展,从而可能为治疗PE提供新的靶点和治疗策略。
1.1.5 Cbl介导Met泛素化降解 Cbl(casitas B-lineage lymphoma, Cbl)属于RING家族E3 泛素连接酶,通过介导多种受体的泛素化和降解参与免疫调节[17]、Wnt/β-catenin信号转导[18]或血管内皮生长因子信号转导[19]等细胞生命过程。
肝细胞生长因子(hepatocyte growth factor, HGF)是胎盘发育的重要调节因子。一旦配体HGF与其受体Met结合,就会发生二聚化和磷酸化,激活MEK/Erk、PI3K等多种下游信号通路,通过影响滋养细胞迁移、侵袭和血管重塑,调节滋养层功能[20]。Li等[21]的研究显示,HGF与Met的结合触发了Met的磷酸化和下游的Erk信号级联,并启动了窖蛋白-1(Caveolin-1, CAV-1)耦合的内吞作用以及Cbl介导的Met的泛素化降解,从而调控滋养细胞的分化。而滋养细胞长期缺氧会诱导Met通过CAV-1耦合作用导致过度内吞,并阻碍Met的蛋白酶体降解,共同导致Met蛋白在细胞内的累积增加,使得HGF/Met信号无法调节滋养细胞侵袭,进一步由于滋养细胞对子宫螺旋动脉的重塑受限而加剧胎盘缺氧,从而形成恶性循环,最终可能导致早发型PE的发生。Rahman等[22]的研究设计了一种新型串联泛素结合模体(ubiquitin-interaction motif, UIM)肽,通过竞争性抑制内吞作用相关蛋白epsin与血管内皮生长因子受体2的UIM结构域依赖性结合,促进了c-Cbl介导的epsin泛素化,在调节血管生成障碍性疾病的治疗中发挥作用,为治疗PE提供了潜在靶点。
1.1.6 CRL3介导底物蛋白泛素化 Cullin3-RING连接酶(Cullin3-RING ligase, CRL3)属于Cullin-RING连接酶超家族,是最常见的一类E3泛素连接酶[23],包含Cullin3支架蛋白、RBX1环指蛋白和含有BTB结构域的底物适配蛋白,识别并招募底物蛋白参与泛素化降解过程[24]。Zhang等[25]的研究发现,CUL3的丰度和nedd化水平,以及RhoBTB1、KLHL2等CUL3的底物适配蛋白在PE胎盘螺旋动脉中表达明显减少;而CRL3的底物,包括PDE5、WNK3和RhoA表达增加。其中,PDE5的累积损害血管舒张;而RhoA与WNK3和WNK1共同作用,促进血管收缩并诱导血管平滑肌细胞的增殖[26]。表明CUL3-KLHL2-WNK3/WNK1和CUL3-Rho-BTB1-PDE5信号的功能障碍破坏了血管收缩和血管扩张的平衡,导致PE胎盘螺旋动脉的异常重塑,从而引起胎盘血流减少、促进胎盘缺血缺氧,促进了PE患者的进行性高血压。
临床研究表明,PE患者易出现水钠潴留[27],表明肾小管功能障碍可能促进PE中高血压的发展。Zhang等[25]的进一步研究发现,PE小鼠肾脏中CUL3的丰度和nedd化水平下降,其在肾脏表达的特异性底物适配蛋白KLHL3水平同样降低。升高的底物蛋白WNK4、WNK1磷酸化并激活下游SPAK/OSR1和噻嗪类敏感的Na-Cl协同转运蛋白(NCC)并且损害血管舒张功能,证明远端肾小管的WNK激酶激活同样促进PE的高血压,而血管和肾脏远曲小管结构重塑可能有助于解释为什么PE患者远期较大的心血管疾病和肾脏疾病风险[28]。而质子泵抑制剂治疗可以部分减少PE小鼠模型中PDE5、WNK激酶和RhoA/ROCK活性的积累,强调了质子泵抑制剂治疗PE的可能机制以及应用前景。
1.1.7 MULE介导p53、Mcl-1泛素化降解MULE(Mcl-1 ubiquitin ligases E3)是一个特异性调节髓细胞白血病因子1(myeloid cell leukemia sequence 1, Mcl-1)蛋白稳定性的E3泛素连接酶,属于HECT家族E3泛素连接酶。
Mcl-1是B淋巴细胞瘤-2(B-cell lymphoma-2, Bcl-2)家族蛋白中重要的抗凋亡蛋白成员,在多种恶性肿瘤(急性细胞性白血病、多发性骨髓瘤等)中呈高表达,通过与p53等促凋亡蛋白相互作用,抑制肿瘤细胞凋亡。而在PE中,缺氧环境使得MULE表达受到低氧诱导因子-1α(hypoxia inducible factor-1α, HIF-1α)和转化生长因子-β3影响而增加,上调的MULE优先结合并降解p53。由于p53可通过Bcl-2家族蛋白影响凋亡,因此p53表达减少引起促凋亡的Mcl-1可变剪接体Mcl-1c表达增加[29],导致滋养细胞过度凋亡而使滋养细胞浸润能力减弱,引起胎盘浅着床和血管重塑障碍,继而促进PE的发生。
1.2 其他参与PE发病机制泛素化修饰的分子
由于E3泛素连接酶在PE发病机制中作用的研究尚处于早期阶段,目前的研究提出了一些可能影响PE发病机制的泛素化途径。Yuan等[30]的研究表明,缺氧时HIF-1α表达增加导致E3泛素连接酶斑点型锌指结构蛋白表达增加,可能通过靶向PI3K/AKT/GSK-3β通路抑制滋养细胞的浸润,从而导致PE的发生;Zhao等[31]的研究假设,PE胎盘中泛素特异性蛋白14的上调可能通过去泛素化过程激活NF-κB,从而上调胎盘中促炎症细胞因子的表达,通过调节炎症反应来促进PE的进展;Li等[32]的研究发现,PE胎盘滋养细胞中泛素特异性蛋白5呈现低表达,下调了β-catenin及其下游信号c-Myc和Cyclin D1的表达从而抑制滋养细胞的增殖;Yang等[33]的研究发现,PE胎盘中单克隆非特异性抑制因子β表达减少,通过蛋白酶体降解胰岛素样生长因子2抑制滋养细胞侵袭[33]。
2 小泛素样修饰与PE发病机制
2.1 小泛素样修饰概述
各种PTM并不是孤立的,彼此之间存在交互应答和相互作用,其中泛素化修饰与类泛素化修饰之间关系密切。小泛素蛋白样修饰蛋白(small ubiquitin-related modifier proteins, SUMO)作为其中一种类泛素修饰因子,与泛素分子的二、三级结构以及酶级联的修饰过程极其相似,但两者的作用不尽相同。泛素化修饰主要介导底物蛋白降解,而SUMO化、去SUMO化修饰则主要在SUMO分子以及SUMO特异性蛋白(sentrin specific protease, SENPs)的调节下[34],通过增强底物蛋白的稳定性,在蛋白质相互作用等方面参与细胞生命过程的调控[35]。哺乳动物表达SUMO1-4这4种SUMO蛋白同种型,其中SUMO-2和SUMO-3由于高序列相似性(97%)而经常被描述为SUMO-2/3。
2.2 PE胎盘中SUMO化修饰水平增加
分子机制导致滋养细胞发育异常引起胎盘功能障碍与产科并发症有关[36]。研究显示,通过影响蛋白质稳定性、活性和细胞内定位,SUMO化修饰通过引起胎盘转录因子GCM1[37]、下游调控元件拮抗分子[38]、p53[39]以及HIF-1α[40]等的表达变化调节滋养细胞分化,而这些转录因子经证明与PE的发病机制密切相关[41-43]。
Baczyk等[44]的研究发现,胎盘中SUMO蛋白的表达呈现出时空差异。SUMO-1和SUMO-4在孕早期定位于细胞滋养层,并随着妊娠的发展迁移至合体滋养层;相反,SUMO-2/3在整个妊娠期间均匀分布在整个细胞滋养层和合体滋养层中。而在模拟PE胎盘的氧化应激条件下,SUMO-1和SUMO-4的SUMO化水平显著增加、并且细胞质中SUMO-1和SUMO-4蛋白的表达增加;而SUMO-2/3则易位到细胞核,其生物利用度和转录活性受到抑制。Baczyk等[42,45]的研究结果证明SUMO化修饰在正常人类胎盘发育起着重要作用,而各种细胞应激源能够在胎盘中诱导SUMO蛋白在细胞内、外的差异分布,导致PE胎盘表现出过度SUMO化,从而表明了SUMO化修饰的动态调节与PE等疾病呈现的胎盘功能障碍密切相关。下面将阐述参与PE发病机制SUMO化修饰的具体分子。
2.2.1 Ct-7的SUMO化修饰 Snider等[46]的研究表明特别是在氧化应激的环境下,SUMO化修饰在调节细胞骨架蛋白中起着非常重要的作用。而Baczyk等[44]通过质谱分析发现,人胎盘JAR细胞中最丰富的SUMO化靶标属于角蛋白家族。在进一步分析蛋白质相互作用的研究中证实,相对于SUMO-2/3而言,人胎盘JAR细胞在氧化应激以及炎症情况下,SUMO-1、SUMO-4与细胞角蛋白-7(cytokeratin-7, Ct-7)之间的相互作用明显增加,进一步证实了Ct-7是胎盘SUMO化修饰的靶标。
Riquelme等[47]的研究显示,人PE胎盘显示出50%的Ct-7表达下调,认为PE胎盘中Ct-7表达水平下调引起滋养层细胞骨架变弱,从而导致滋养层碎片更多进入母体循环中,显示出PE胎盘滋养细胞凋亡和坏死物质脱落增加的特点。而Baczyk等[44]的研究数据表明,在人类胎盘中,Ct-7的SUMO化修饰损害细胞骨架丝的稳定性,可能与滋养层细胞骨架重塑有关,表明Ct-7的SUMO化修饰与PE胎盘功能障碍相关。
2.2.2 HIF-1α的SUMO化修饰 HIF-1α作为缺氧敏感调节因子,通过调节血管内皮生长因子(vascular endothelial growth factor-A, VEGF)、可溶性血管内皮细胞生长因子受体-1(soluble vascular endothelial growth factor receptor-1, sFlt-1)等靶基因的表达,在PE胎盘中表达异常升高并抑制滋养细胞增殖和浸润能力,导致胎盘浅着床、血管重塑障碍而引起PE。Bhattacharjee等[48]的研究证明,PE胎盘缺氧、氧化应激的环境使得HIF-1α在常氧情况下经SUMO化而发生的羟化以及泛素化降解受到抑制,同时SENP3转位激活并介导HIF-1α进行去SUMO化,使HIF-1α在PE胎盘中呈现稳定性高表达,进一步调控VEGF、sFlt-1的表达并形成恶性循环,引起一系列PE的相关症状。
2.2.3 SATB1的SUMO化修饰 特异AT序列结合蛋白1(special AT-rich sequence-binding protein 1, SATB1)已经被证实可促进多种恶性肿瘤细胞的侵袭和转移[49],最近的研究还表明SATB1参与了胚胎的分化和发育。Rao等[50]的研究发现,从妊娠早期开始SATB1在胎盘的高侵袭性滋养层中呈高表达,并随着妊娠的进展逐渐减少,提示SATB1与滋养层功能的调节之间存在相关性。此外在PE胎盘中滋养细胞的迁移和侵袭潜能明显降低,并伴有SATB1的表达显着下降,证实了下调的SATB1表达与滋养层功能受损有关。
以往的研究表明SATB1蛋白的生物学功能影响Wnt/β-catenin信号通路[51]。Wnt/β-catenin信号通路已被证明参与调控滋养细胞的功能,其中β-catenin的表达在PE胎盘中显着降低。Rao等[52]的研究发现,氧化应激诱导SATB1在lysine-744处的SUMO化修饰并降低了SATB1的蛋白水平及其稳定性;而较高的SATB1水平则增加了β-catenin水平,减少了活性氧(ROS)累积以及细胞凋亡,同时促进了滋养细胞迁移和侵袭。证实了PE胎盘氧化应激的情况诱导了SATB1蛋白的SUMO化修饰,通过抑制Wnt/β-catenin信号通路损害滋养细胞迁移和侵袭能力。其中,使用WNT/β-catenin信号通路特异性抑制剂DKK1减弱了上调的SATB1表达对受损滋养细胞的恢复作用,提示靶向SATB1和Wnt/β-catenin信号通路对于治疗PE的潜在机制。
3 结 语
PE是妊娠期特有的常见并发症,泛素化修饰和类泛素化修饰中的小泛素样修饰在PE的发病机制中起重要作用。作为泛素化修饰的重要酶,包括RING家族的SCFFBW2、SCFβ-TrCP、CCNG2、Cbl、CRL3以及HECT家族的MULE等在内的E3泛素连接酶在不同方面影响PE发病机制;而类泛素化修饰中的小泛素样修饰则主要通过Ct-7、HIF-1α、SATB1等蛋白经SUMO化、去SUMO化修饰影响PE发病机制的进展,并为PE治疗提供了潜在靶点和治疗策略。目前泛素化修饰与类泛素化修饰在PE发病机制中的研究尚处于早期阶段,有待进一步对于参与PE发病机制的泛素化和类泛素化分子进行研究,并且利用相关分子的修饰机制开发针对修复滋养细胞损伤的小分子药物治疗PE。
