镁调控心血管钙化机制的研究进展
2023-01-13吴昊刘晓红
吴昊 刘晓红
随着老龄化的加剧,心血管疾病的发病率急剧上升,是世界范围内导致死亡的主要原因之一[1]。血管和瓣膜钙化是心血管疾病的高风险因素,通常预示着心血管疾病患者的不良预后[2]。关于血管钙化和瓣膜钙化的原因曾被认为是一种退行性变,随着研究的深入,发现钙化是一个很复杂的过程,涉及矿物质的沉淀、内皮细胞功能障碍、成骨细胞转分化,还涉及严格调控的细胞介导过程。当细胞外液的磷酸盐和钙离子过多时,会形成磷酸钙晶体并沉积在血管壁、心脏瓣膜等组织中,导致血管和瓣膜弹性减小、形成狭窄,但具体机制尚不明确,目前仍无有效的预防和治疗手段。心血管钙化常见的类型主要有主动脉内膜钙化和瓣膜钙化,二者的发病原因可能不尽相同,但仍有不少相似之处[3]。血管内膜钙化与动脉粥样硬化密切相关,通常由高脂血症引起,血清低密度脂蛋白(low density lipoprotein,LDL)在内皮下腔聚集,招募单核细胞和巨噬细胞并导致炎症,促进成骨的发生[4]。同样,在瓣膜钙化中,由于高频率高强度的震荡流动可产生利于异常物质沉积的环境,而脂蛋白、脂质、炎症细胞的堆积均是钙化性心脏瓣膜病的发病机制[5]。
1 镁离子在心血管钙化中的作用
镁是人体重要的阳离子,机体约60%的镁存在于骨骼中,其余大部分在骨骼肌及其他组织器官细胞内,仅有1%~2%在细胞外液中。镁是人体内多种代谢反应的辅助因子,参与调节各种离子通道的电流,同时在调控细胞生长,维持心肌、骨骼肌兴奋性等方面具有重要作用。近年来,人们发现镁参与调节心血管功能的许多重要过程,镁的稳态失衡会导致高血压、心律失常、动脉粥样硬化、动脉钙化、血管老化等一系列心血管疾病[6]。在以往的研究中镁被认为是天然的生理钙阻滞剂,镁缺乏似乎会促进血管钙化[7]。镁补充剂在人类和实验动物研究中已被证实可抑制成骨基因的表达,因此,成骨基因被认为是镁的作用靶点从而阻止钙化的发生。也有实验证明镁可以通过影响钙磷代谢、脂质沉积、氧化应激、炎症反应等过程调节钙化进展,但其抑制血管和瓣膜钙化的具体机制目前仍不清晰[8]。
2 镁与血管和瓣膜钙化
2.1 促进钙磷代谢稳态
血管和瓣膜钙化的初期通常是由于血液中Ca2+、磷酸盐和血清蛋白过度沉积形成无定形的可溶性钙蛋白单体,随后这些单体聚集形成初级钙蛋白颗粒(calciprotein particle 1,CPP1)。CPP1分子量较小,最初无毒无害,但随着胎球蛋白A(fetuin A)、骨桥蛋白(osteopontin,OPN)等大分子钙化抑制蛋白表达的减少,CPP1逐渐成熟为次级钙蛋白颗粒(CPP2)。CPP2分子量则相对较大,由蛋白质覆盖羟基磷灰石组成。CPP2是血管平滑肌细胞(vascular smooth muscle cell,VSMC)转分化和血管钙化的重要驱动因素,已被证实可在体外诱导VSMC中runt相关转录因子2(runt-related transcription factor 2,Runx2)、碱性磷酸酶(alkaline phosphatase,ALP)、成骨细胞特异性转录因子(osterix,Osx)等成骨蛋白的表达上调[9]。镁可以在此级联反应中发挥抑制作用,在早期阻断CPP1向CPP2的转变,降低成骨基因的表达[10]。羟基磷灰石是钙化组织中最丰富的晶体类型,镁可以取代羟基磷灰石中的Ca2+,形成含Mg2+的白磷灰石,降低了致病性并增加溶解度,从而减少羟基磷灰石的沉积。 镁虽然能有效阻止CPP1向CPP2进展,但并不能阻止CPP1的形成,且在CPP2形成后无法逆转已经形成的钙化。镁通过改变钙稳态发挥一定的保护作用,但不会改变血管和瓣膜的生长、成分或结构。
2.2 抑制脂质沉积
血管和瓣膜钙化的早期,高胆固醇血症、脂蛋白(lipoprotein,Lp)a和LDL水平异常等均是相关危险因素[11]。脂蛋白在血管和瓣膜的内皮下腔聚集,在细胞代谢产物的作用下发生轻度氧化等非酶修饰,轻度氧化的磷脂是钙化生物活性因子,可在体外促进钙化。研究表明,血清镁浓度降低与人体脂质代谢紊乱之间存在密切关联,镁可以通过调节脂质代谢相关转录基因酶的活性和表达量参与控制脂质的生成和分解[12]。镁能竞争性抑制内源性胆固醇合成限速酶(3-hydroxy-3-methylglutaryl-coenzyme A,HMG-CoA)还原酶的磷酸化,激活卵磷脂胆固醇脂酰转移酶(lecithin-cholesterol acyltransferase,LCAT),阻断羟甲戊酸代谢途径,通过降低LDL控制胆固醇的生物合成[13]。氧化LDL(oxidized LDL,ox-LDL)可以促进脂蛋白相关磷脂酶A2(lipoprotein-associated phospholipase A2,Lp-PLA2)的表达,Lp-PLA2能增加溶血磷脂酰胆碱( lyso-phosphatidylcholine,LPC)通过PKA信号通路激活钙化[14]。Fazlali等[15]实验证明补充镁补充可改善血脂,降低ox-LDL并下调凝集素样低密度脂蛋白受体-1(lectin-like oxidized low-density lipoprotein receptor-1,LOX-1)。有研究表明,氧化的脂质会通过骨形态发生蛋白2 (bone morphogenetic protein 2,BMP2) 介导瓣膜钙化,而缺乏脂蛋白可抑制钙化的发生[16]。镁有助于上调脂蛋白脂肪酶活性,这种酶可以促进生成高密度脂蛋白(high density lipoprotein,HDL),而HDL是ALP表达的抑制剂,减少镁摄入会增加极低密度脂蛋白(very LDL,VLDL)和LDL,并降低HDL。因此,维持脂质代谢平衡可能是镁抑制血管和瓣膜钙化的一种潜在途径。
2.3 抑制氧化应激
氧化应激是血管和瓣膜钙化的重要病理因素之一[17]。当机体活性氧ROS生成增多超过了细胞的抗氧化能力时,会刺激细胞发生表型转分化,上调成骨分化相关转录因子RUNX2的表达,促进钙化的发生[18]。外源性H2O2可以通过IP3/AKT途径上调核结合因子 α1(Cbfa1)的表达,促使ALP表达上调,并促进成骨细胞表型转分化。还原型辅酶Ⅱ(NADPH)氧化酶是一些非吞噬细胞活性氧(reactive oxygen species,ROS)合成的主要酶体,其诱导生成的ROS可进一步促进其他来源的ROS生成。细胞内镁缺乏已被证实会通过改变耦合呼吸和下调电子传递链,增加ROS的生成,抑制抗氧化,导致由ATP减少引起的NADPH氧化酶活性增加[19]。另一方面,补充镁后过量线粒体ROS生成被抑制,Bcl-2表达升高,Bax等表达降低,通过抑制低氧诱导因子1(hypoxia-inducible factor 1,HIF-1)α和p38/JNK信号通路,下调自噬[20]。这表明镁可以通过调控氧化应激抑制血管和瓣膜钙化。
2.4 抑制炎症反应
正常的心血管组织中不存在炎性细胞,而钙化的血管和瓣膜中可观察到明显的炎性改变,浸润的炎性细胞可释放大量的细胞因子和生长因子,通过激活Notch、BMP、Wnt和核因子κB(nuclear factor kappa-B,NF-κB)等信号通路促使钙化的发生[21]。炎症因子可以介导BMP2等促钙化细胞因子的生成及释放,还能下调基质Gla蛋白(matrix Gla protein,MGP)等钙化抑制蛋白的表达,诱导钙磷结晶形成,进一步导致异位钙化的发生。TRPM7是主要的Mg2+通道受体,参与细胞内镁的调节、细胞的粘附和迁移等重要生理过程,同时具备蛋白激酶和离子通道双重结构,广泛存在于机体组织中[22]。TRPM7激酶结构缺失的细胞,表达促炎表型,促进炎症细胞的粘附以及环氧合酶和纤溶酶原激活物抑制剂的合成。Rios等[23]发现TRPM7激酶结构缺失小鼠模型表现出心血管炎症和纤维化,大量炎性细胞因子、巨噬细胞活化异常,中性粒细胞/内皮细胞黏附增加等,补充镁后可改善该状况,证明了TRPM7缺失而引发的炎症反应和纤维化反应具有镁依赖性,其他相关镁转运蛋白家族也参与了炎症的调节。近年来,镁与炎症之间的高关联度被广泛报道,血浆镁水平与单核细胞趋化蛋白1(monocyte chemotactic protein-1,MCP-1)、C反应蛋白(C-reactive protein,CRP)和肿瘤坏死因子-α(tumor necrosis factor α,TNF-α)呈负相关,而镁含量升高可以抑制炎症反应[24]。除此之外,低镁可以通过激活肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system,RAAS)激活炎症,增加Ca2+通道开放、活化吞噬细胞,激活NF-κB信号通路,促进细胞因子和促炎因子的转录[6]。因此,镁可以通过抑制炎症反应减缓血管和瓣膜钙化的发生发展。
2.5 抑制成骨细胞样表型转分化
正常的VSMCs和瓣膜间质细胞(valve interstitial cells,VICs)通常都表现为静止的表型,但在一些刺激因素下会向成骨细胞样表型转分化,促使多种钙化因子表达量增加,导致血管和瓣膜增厚变硬,最终发生钙化[25]。Wnt/β-Catenin通路在介导血管和瓣膜钙化过程中发挥重要作用,不但能促进Runx2的表达,还能诱导基质金属蛋白酶2/9(matrix metalloproteinase-2/9,MMP-2/9)的生成,是成骨细胞转分化的重要信号通路[26]。镁可以通过TRPM7抑制Wnt/β-Catenin通路阻止成骨细胞样表型转分化和钙化的发生,非选择性TRPM7阻断剂2-氨基乙氧基硼酸二苯酯(2-aminoethoxydiphenylborate,2-APB)可以使镁对成骨标志物表达和钙化的保护作用明显被削弱[27]。研究发现镁可以通过稳定Ca2+感应受体CaSR的活性,维持MGP的合成来防止成骨细胞样表型转分化导致的钙化[28]。研究证实镁可抑制高磷酸盐诱导激活的Wnt/β-Catenin通路,降低成骨转录因子Cbfa-1及其下游因子osterix的表达,上调钙化抑制剂MGP和骨保护素(osteoprotegerin,OPG)的表达。更有发现,在钙化已经建立后添加镁,钙化不仅会停止,甚至会减少,并伴随着 Cbfa-1、osterix、MGP和OPG基因表达量的显著改变[29]。因此,镁抑制成骨细胞样表型转分化是实现血管和瓣膜抗钙化作用的一种潜在机制。
2.6 维持内皮细胞屏障功能
内皮细胞位于血液和血管或瓣膜之间的交界面,直接接触血液,通过调节渗透、炎症以及血栓的形成保护组织结构[30]。内皮细胞可以快速感受到血流动力学以及血液成分的病理改变等异常刺激,并引起相应变化,在血管和瓣膜的钙化过程中至关重要。近期有研究表明,内皮细胞自身可通过内皮-间质转分化(endothelial-mesenchymal transition,EndMT)去分化成为间质细胞,获得多分化潜能后为钙化的发生提供了细胞基础[31]。内皮细胞可以通过释放相关信号分子和细胞因子等刺激成骨样转分化,并分泌胞外囊泡为钙化提供成核位点[32]。既往研究表明,血清镁浓度与内皮功能呈负相关,低镁会影响内皮屏障完整性和稳定性,并进一步影响相关组织的通透性、张力等性质[33]。镁可以通过TRPM7和镁离子转运蛋白(magnesium transporter 1,MagT1)调节内皮细胞的活性以及增殖能力,进而调整内皮屏障的完整性[34]。细胞外镁浓度增加可以与部分Ca2+结合位点结合,减少细胞膜表面的Ca2+-Na+交换从而减少细胞膜的内向电流,轻度增加外向电流造成细胞膜超极化,使细胞膜状态更加稳定。PI3K/Akt 通路有助于各种病理条件下内皮细胞的存活并有助于激活Nrf2,Nrf2有调节内皮细胞的作用,镁通过PI3K/Akt 信号通路可能是一种潜在的保护机制[35]。可见,镁可以通过维持内皮细胞屏障产生抑制血管和瓣膜钙化的作用(图1)。
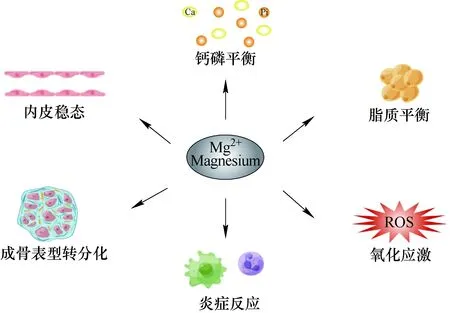
镁可以通过维持钙磷代谢平衡、抑制脂质异常沉积、降低氧化应激水平、抑制炎症反应、抑制成骨细胞样表型转分化和维持内皮细胞功能稳态,进而延缓或减轻血管和瓣膜的钙化图1 镁在心血管钙化中的作用
3 展望与总结
血管和瓣膜的钙化在临床上尚无有效的预防和治疗方法,阐明其发生的具体机制,寻找一个合适的干预靶点有着十分重要的临床意义。目前越来越多的证据表明,镁可以通过多种途径直接或间接参与血管和瓣膜钙化的过程,并改善钙化倾向,而且各个机制之间存在相互交叉的作用。因此,镁的多途径作用特征使其有望成为一种钙化防治的新方法。然而,关于镁抑制钙化的具体机制、细胞通路等的研究仍然十分有限,未来应该就此方面开展深入的研究,补镁可能延缓或减轻血管和瓣膜钙化的进展,成为一个全新可靠的预防及治疗靶点。
利益冲突:无
