斑马鱼gpr112a基因敲除品系的构建
2023-01-11孙鲁宁杨博宇朱俊伟杨添乐谢华平
孙鲁宁,杨博宇,刘 玲,朱俊伟,杨添乐,彭 政,郑 澜,谢华平,d∗
(湖南师范大学 a.动物肠道功能调控湖南省重点实验室;b.动物营养与人体健康实验室;c.体适能与运动康复湖南省重点实验室;d.淡水鱼类发育生物学国家重点实验室,长沙 410081)
G蛋白偶联受体(G protein coupled receptors,GPCRs)是构成人体的最大最普遍的膜受体家族,具有大约800个不同的成员,可识别多种细胞外配体,如核苷酸、肽、脂质和蛋白质,并将其信号转导到细胞中,是药物治疗最常见的靶点[1-3]。GPCRs均具有一个基本相同的分子结构:7个由25~35个连续残基组成的跨膜疏水结构域。此外,它们还具有共同的信号传导机制,即其与普遍存在的鸟嘌呤核苷酸结合调节蛋白(G蛋白)相互作用,以调节细胞内第二信使的合成[4]。黏附类G蛋白偶联受体(adhesion G protein coupled receptors,aGPCRs)是GPCRs的5个主要家族中的一族,其中“黏附”一词反映了其在细胞黏附中的潜在作用[5-6]。GPR112(ADGRG4)是aGPCRs家族的一员,由GPR112基因编码。目前国际上对GPR112的针对性的研究非常少,其生物功能也不为人知。作为其为数不多的研究成果之一,GPR112曾被确认为神经内分泌癌细胞的标记基因[7]。
斑马鱼(Daniorerio)作为脊椎动物模式生物具有许多优点,包括其透明的胚胎便于观察细胞和器官的形态以及报告基因的表达,且CRISPR-Cas9基因编辑技术已被熟练地运用于斑马鱼的基因组编辑中[8-9]。斑马鱼gpr112a(adgrg4a)基因是人类GPR112的直系同源基因,经高通量实时定量PCR鉴定,其最早在受精后5 dpf(days post fertilization)出现显著表达,于11 dpf出现表达量的峰值,且在成鱼肠道中高表达[10]。
我们首次使用CRISPR-Cas9基因编辑技术对斑马鱼的gpr112a基因进行特异性敲除,以获得gpr112a基因缺陷的突变体斑马鱼,从而进一步研究gpr112a基因缺失对斑马鱼的生长发育造成的影响以及其生物学功能。
1 材料与方法
1.1 材料
TU品系斑马鱼来自国家斑马鱼资源中心(China Zebrafish Resource Center,CZRC),于本实验室养殖。
研究所用主要试剂和耗材包括:E3 Buffer(5.00 mmol/L NaCl、0.17 mmol/L KCl、0.33 mmol/L CaCl2、0.33 mmol/L MgSO4[11],均购自国药集团化学试剂有限公司);斑马鱼养殖与繁育系统(北京爱生科技发展有限公司);Rapid Taq Master Mix(南京诺唯赞生物科技有限公司);柱式DNA胶回收试剂盒(生工生物工程股份有限公司);DL2000 DNA Marker[宝生物工程(大连)有限公司];T7转录试剂盒(Promega公司,美国);RNeasy Mini Kit(Qiagen,德国);Lysis Buffer(50 mmol/L Tris-HCl pH=8.0,25 mmol/L EDTA,100 mmol/L NaCl,0.5% SDS,100 μg/mL Proteinase-K,其中Tris和HCl购自国药集团化学试剂有限公司,EDTA和SDS购自生工生物工程股份有限公司,Proteinase-K购自北京全式金生物技术股份有限公司);pMD18-T Vector Cloning Kit[宝生物工程(大连)有限公司];Plasmid Mini-Prep Kit(北京全式金生物技术股份有限公司);4%多聚甲醛(上海源叶生物科技有限公司);磷酸盐缓冲液(武汉赛维尔生物科技有限公司);低熔点琼脂糖[西格玛奥德里奇(上海)贸易有限公司]。
Sanger测序委托北京擎科生物科技有限公司进行。
1.2 斑马鱼的饲养与繁殖
在28℃恒定温度下养殖成年斑马鱼,并保持14 h光照和10 h黑暗的明暗循环。性成熟的雌雄斑马鱼各1条,每周产卵1次;收集胚胎放至E3 Buffer中,每12 h换1次E3 Buffer,饲养至5 dpf开始喂食草履虫。当斑马鱼发育到14 dpf之后,将幼鱼放置到斑马鱼养殖与繁育系统中进行养殖,并喂食丰年虫,早中晚共3次。
1.3 sgRNA靶位点的设计
在 Ensembl(http://www.ensembl.org/index.html)网站上查询gpr112a的转录本与内、外显子信息;找出所有紧邻5'-NGG-3'(PAM)的候选靶序列,其大小一般为18~20 bp。sgRNA上游引物序列F的序列构成为:于靶序列前加保护碱基(GCG)和T7启动子(TAATACGACTCACTATA),靶序列后加sgRNA骨架序列的上游序列(GTTTTAGAGGCTAGAAATAGG)。sgRNA下游引物R为固定序列(AAGCACCGACTCGGTGCCACT)。根据gpr112a基因的靶位点,在靶位点的上下游通过Primer3.0(https://bioinfo.ut.ee/primer3-0.4.0/)在线工具分别设计正、反检测引物gpr112a-F和gpr112a-R(表1)。

表1 本研究涉及的引物序列Tab.1 Primer sequences involved in this study
1.4 聚合酶链式反应(PCR)
根据说明书,在200.0 μL离心管中混合10.0 μL Rapid Taq Master Mix、1.0 μL上游引物、1.0 μL下游引物和0.5~1.0 μL模板,加入双蒸水补齐至20.0 μL。将混合好的反应溶液放入聚合酶链式反应(polymerase chain reaction,PCR)热循环仪(Thermo Fisher Scientific公司,美国)中,依照Rapid Taq Master Mix设置常规PCR程序,其中退火温度为58℃,延伸时间为15 s,共30个循环。反应结束后,对反应产物进行琼脂糖凝胶电泳分析并成像。
1.5 显微注射
收集15 min内产的斑马鱼受精卵,将gpr112a基因靶位点的sgRNA1、sgRNA2和Cas9蛋白混匀(终质量浓度分别为50.00、50.00、250.00 ng/μL)注射到斑马鱼受精卵中,每颗胚胎的注射体积控制在1~2 nL之间,随后置于28℃的恒温箱中培养。
1.6 斑马鱼基因组DNA的提取
将60 dpf左右的斑马鱼麻醉,用眼科剪刀剪取约0.2 mm2的尾鳍末端放入离心管中,再加入50 μL Lysis Buffer,55℃水浴12~24 h。水浴结束后,在离心管中加入50 μL预冷的异丙醇,混合均匀后12 000 r/min离心10 min,弃上清。加入100 μL 70%乙醇,振荡混匀,12 000 r/min离心10 min,弃上清。于55℃烘箱干燥10 min后,加入20 μL双蒸水,振荡溶解,得到斑马鱼的基因组DNA溶液。
1.7 gpr112a基因型的鉴定
以单条斑马鱼的基因组DNA为模板进行PCR反应,gpr112a-F、gpr112a-R分别为上游引物和下游引物。根据产物的琼脂糖凝胶电泳图来确定斑马鱼的基因型为野生型、gpr112a基因突变杂合子还是gpr112a基因突变纯合子。
1.8 T-A克隆
在灭菌的离心管种加入 1 μL pMD18-T、5 μL Solution I(来自 pMD18-T Vector Cloning Kit)、100 ng带有A尾的纯化DNA片段,加入双蒸水补齐至10 μL以配置成连接溶液。将含有连接溶液的离心管放入PCR仪中,设置温度为16℃,时间为30 min。反应结束后,将连接溶液加入至100 μL的感受态大肠杆菌中,于冰上放置30 min后42℃热激1 min,再次将其转移至冰上放置2~5 min。在超净工作台中将感受态混合液滴加至含氨苄青霉素钠的肉汤琼脂培养基表面,涂抹均匀后于37℃培养12 h。从培养基表面挑取单克隆菌落加入到肉汤液体培养基中扩培,使用Plasmid MiniPrep Kit提取质粒。
1.9 体视显微镜成像
将斑马鱼幼鱼收集到离心管中,尽量将水吸干,加入1 mL 4%多聚甲醛,于4℃存放过夜。次日,将离心管中的液体吸出,使用磷酸盐缓冲液冲洗幼鱼2次。使用1%低熔点琼脂糖固定幼鱼,使其左侧身体朝上。使用体式显微镜(Leica公司,德国)观察其侧面观并拍照记录。
2 结果与分析
2.1 gpr112a基因敲除sgRNA的合成
为了将野生型斑马鱼的gpr112a基因敲除,我们现合成了一对用于显微注射的sgRNA。以Template DNA[12]为模板,sgRNA1-F/sgRNA-R或sgRNA2-F/sgRNA-R为正向引物和反向引物进行PCR扩增。比照DNA Marker,用洁净的小刀将100 bp左右的特异性条带切下,并按照柱式DNA胶回收试剂盒说明书的要求进行DNA纯化回收。以回收的DNA作为模板,使用T7体外转录试剂盒进行体外转录,获得sgRNA1和sgRNA2。使用RNeasy Mini Kit将两种sgRNA纯化后进行琼脂糖凝胶电泳,验证sgRNA的成功合成(图1)。
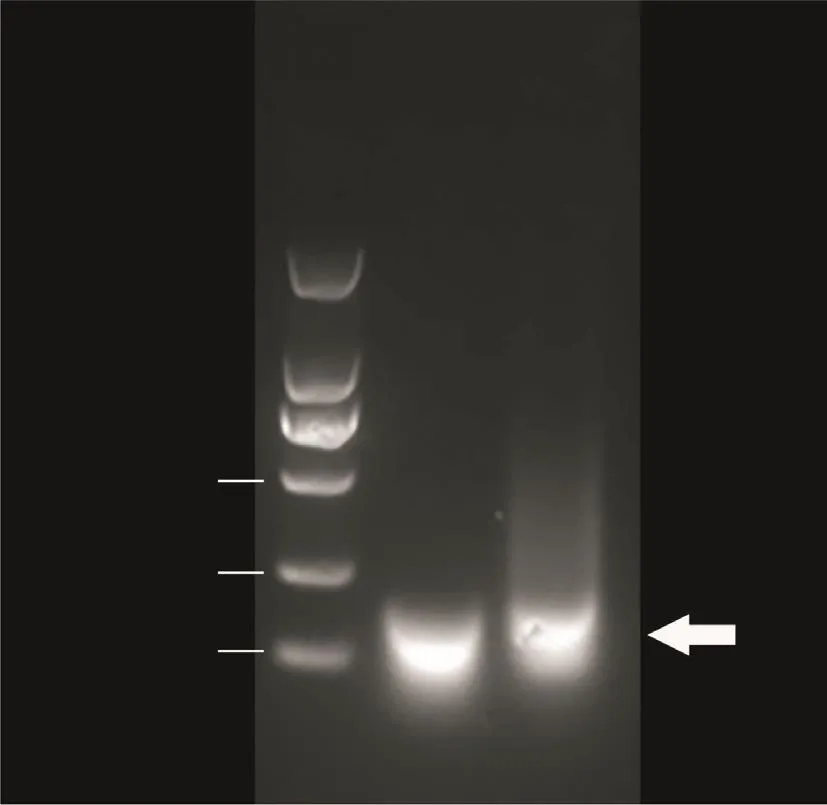
图1 用于gpr112a基因敲除的sgRNA的琼脂糖凝胶电泳Fig.1 Agarose gel electrophoresis of sgRNA for gpr112a knockout
2.2 gpr112a基因敲除的有效性鉴定及F0代突变等位基因嵌合体成年斑马鱼的筛选
为了验证斑马鱼胚胎的显微注射靶位点是否有效,选择注射后发育至60 dpf的斑马鱼尾鳍提取基因组DNA,并将其作为模板,使用基因型鉴定引物(gpr112a-F、gpr112a-R),通过PCR扩增进行注射有效性的鉴定。琼脂糖凝胶电泳结果表明,野生型的PCR产物特异性条带为431 bp,1~7泳道除了有野生型431 bp条带外,都还出现了一条较为明显的小带,说明我们双靶位点的显微注射可能是有效的(图2)。将这7条F0代斑马鱼继续养至性成熟。

图2 经显微注射的gpr112a基因敲除F0代60 dpf斑马鱼基因型鉴定Fig.2 Genotyping of 60 dpf zebrafish in the F0 generation of gpr112a knockout by microinjection
2.3 gpr112a突变等位基因的T-A克隆及Sanger测序分析确立gpr112a基因敲除品系建立
为了验证F0代中可能存在的gpr112a基因突变是否可以遗传至后代,我们将图2中筛选出的第4号成年F0代斑马鱼与野生型斑马鱼杂交,得到F1代胚胎,培育到2 dpf时随机提取胚胎的基因组,进行基因型鉴定。琼脂糖电泳结果显示,其后代产生了gpr112a突变等位基因的杂合子斑马鱼(图3)。
将图3中10号斑马鱼基因组再次使用基因型鉴定引物进行大量扩增,切取产物中的不同于野生型斑马鱼的较小分子量的条带,使用柱式DNA胶回收试剂盒进行DNA纯化回收。将回收的DNA溶液进行T-A克隆,选取阳性克隆质粒进行Sanger测序,与野生型斑马鱼的gpr112a基因进行比对。该斑马鱼的gpr112a突变等位基因DNA在2号外显子共缺失145 bp,致使其开放阅读框改变,并提前终止翻译(图4a、4b)。分析gpr112a突变等位基因所编码的蛋白质,可证明其主要结构域已缺失(图4c)。因此,此F0代斑马鱼嵌合体含有这种所翻译的蛋白质功能失效的gpr112a突变等位基因的体细胞,且这种突变等位基因能够稳定遗传至后代的突变体F1代。此F1代gpr112a突变等位基因杂合子斑马鱼的诞生即代表了gpr112a基因敲除斑马鱼品系的成功建立。
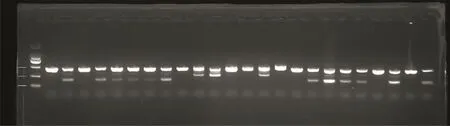
图3 F1代斑马鱼gpr112a突变等位基因杂合子的筛选Fig.3 Screening of heterozygotes for mutant alleles of gpr112a in F1 generation zebrafish
2.4 gpr112a基因突变体纯合子的筛选
为了构建完整的斑马鱼敲除突变体品系,将与上述经过基因测序的F1代斑马鱼同时所产的剩余胚胎培养至性成熟,经基因型鉴定后筛选出gpr112a突变杂合子。将它们的gpr112a突变等位基因T-A克隆后进行Sanger测序,选取测序结果为图4所示的杂合子F1代的雌雄鱼进行自交,提取其后代的基因组DNA进行PCR以鉴定其基因型。琼脂糖凝胶电泳结果显示,此23条gpr112a突变杂合子自交所产的F2代斑马鱼中含9条gpr112a突变纯合子、10条突变杂合子、4条野生型。这证明了gpr112a基因敲除品系建立成功(图5)。
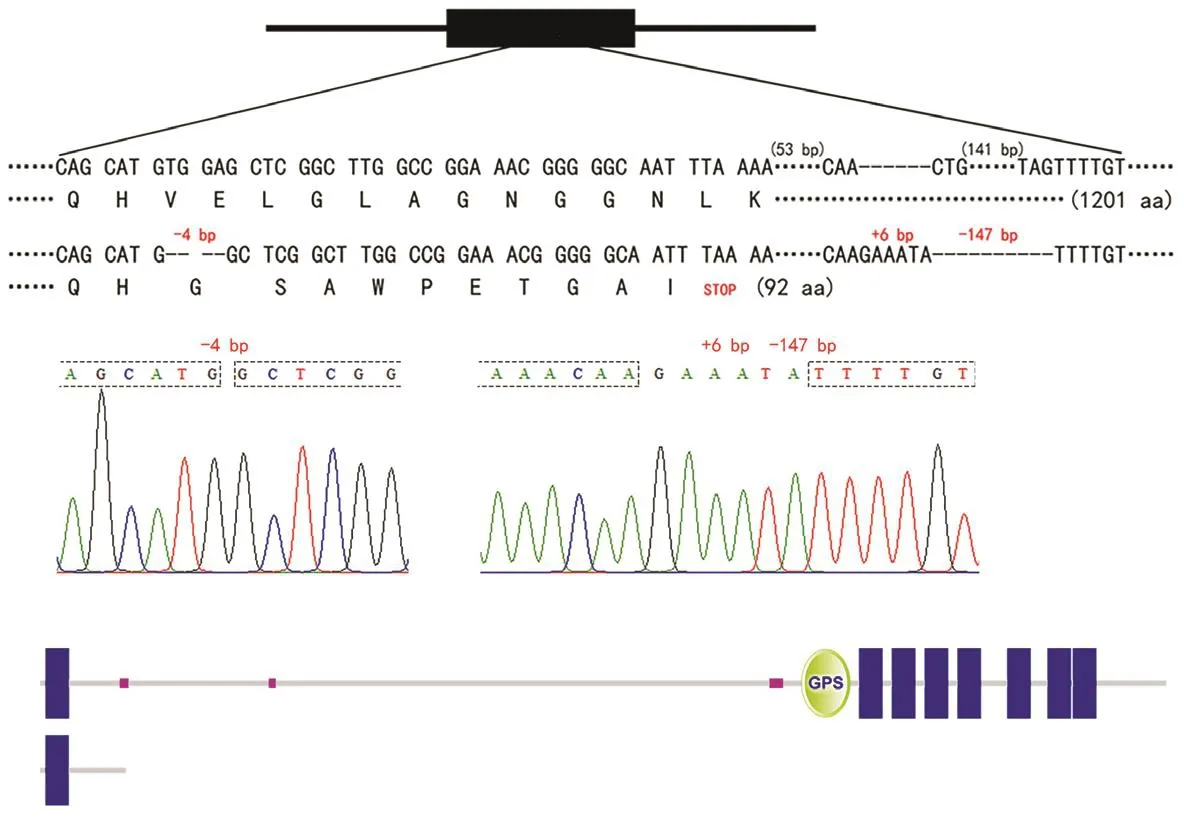
图4 gpr112a突变等位基因序列与其所翻译蛋白质序列分析Fig.4 Sequence analysis of gpr112a mutant allele and its translated protein
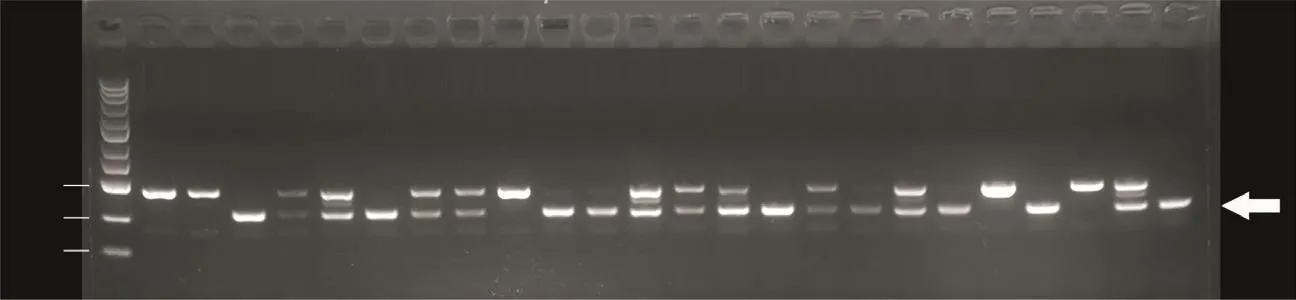
图5 gpr112a突变等位基因纯合子斑马鱼的筛选Fig.5 Screening of zebrafish homozygous for the gpr112a mutant allele
2.5 gpr112a基因突变纯合子斑马鱼相较野生型无明显差异表型
gpr112a突变等位基因纯合子斑马鱼为gpr112a基因表达缺失的斑马鱼。为了观察其与野生型斑马鱼的差异,我们对它进行了体式显微镜白光成像。由于野生型斑马鱼中gpr112a在5 dpf和7 dpf均有显著表达[10],所以我们选取了7 dpf的gpr112a-/-斑马鱼幼鱼,使用体式显微镜拍摄其侧面观。结果显示,gpr112a的缺失并没有致使斑马鱼在受精后7 dpf与野生型出现明显的外观形态差异(图6)。
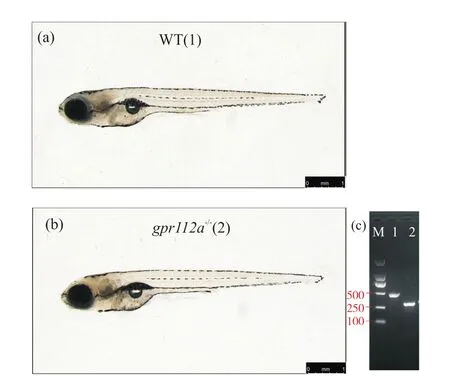
图6 野生型与gpr112a纯合子突变斑马鱼7 dpf侧面观Fig.6 7 dpf lateral view of zebrafish homozygous for wild-type and gpr112a mutant alleles
对同一天受精的野生型和gpr112a-/-斑马鱼持续饲养并观察,直至240 dpf。相较野生型斑马鱼,gpr112a-/-斑马鱼均并没有出现明显的异常表型(图7)。

图7 野生型与gpr112a突变等位基因纯合子斑马鱼240 dpf侧面观Fig.7 240 dpf side view of zebrafish homozygous for wild-type and gpr112a mutant alleles
3 讨论
CRISPR-Cas9作为基因编辑工具,其在生命科学研究中的广泛应用已深刻地影响了科学界,尤其是分子生物学领域[13-14]。CRISPR-Cas9所介导的基因敲除作为基因编辑的一种,其作用机制为RNA引导的Cas9核酸酶可以精确靶向,在基因组中的特定位点诱导DNA双链断裂(double-strand breaks,DSB),触发修复机制,而这种修复所介导的突变有可能会降低或消除基因功能[15-17]。基因敲除的设计与作用方式是灵活多样的,我们所采用的是设计用于靶向gpr112a基因中单个外显子的两个不同位点的sgRNA,将它们与Cas9内切酶混合后同时注射入斑马鱼的受精卵中。这样即可在斑马鱼基因组DNA中引入两个并发的DSB,并导致两个位点之间基因组片段的靶向缺失[18-19]。之后筛选出嵌合体后代中gpr112a的外显子核苷酸缺失数量为非三整数倍的个体,即产生了gpr112a翻译区移码突变的斑马鱼,其为gpr112a蛋白翻译失效的特异性基因敲除品系。其杂合子自交所得的F2代的基因型符合孟德尔分离定律,即野生型和纯合子各约占1/4,杂合子约占1/2。我们基于此研究首次报道了在模式生物中敲除人类GPR112的直系同源基因的方式及结果。
在生物体内存在一些补偿机制致使敲除生物体中的某个基因可能几乎没有表型影响,包括重复基因的存在、代谢途径和调节网络的代偿等[20]。事实上,GPR112在斑马鱼基因组中存在两个旁系同源基因,即gpr112a和gpr112b[10]。旁系同源基因是通过基因的复制而产生的基因[21-22],旁系同源物之间的功能重叠使它们能够补偿彼此的缺失[23-24]。也就是说,旁系同源基因可以互相作为“功能备份”,当一个基因由于突变或者表达受抑制导致功能受损时,它的旁系基因可能就充当替补队员来部分弥补其缺失的功能,维持斑马鱼生理状态的相对正常。另外,随着反向遗传工具的使用,许多报道指出,基因敲除与基因敲低在各种模式生物中出现表型差异[25-27]。由此,遗传补偿或转录适应的概念被提出,但人们目前对它的具体分子机制知之甚少[28-29]。总之,上述机制均是生命体遗传稳健性的体现,我们在斑马鱼gpr112a突变等位基因纯合子上没有观察到与野生型有明显差异的表型,可能是上述多种因素的复合结果。我们未来拟敲除它的旁系同源基因gpr112b,并构建gpr112a、gpr112b双基因敲除的斑马鱼品系,以消除它们可能存在的代偿机制,更好地揭示GPR112在遗传和发育中的作用。
