红花不同花色变异类型叶绿体基因组特征与系统进化分析
2023-01-10丁怡宁毕光耀胡赛文李贺敏
丁怡宁,毕光耀,胡赛文,周 艳,李贺敏,夏 至
红花不同花色变异类型叶绿体基因组特征与系统进化分析
丁怡宁,毕光耀,胡赛文,周 艳,李贺敏*,夏 至*
河南农业大学农学院,河南 郑州 450046
以红花不同花色变异类型为材料,比较分析其叶绿体基因组结构及其与近缘类群的系统发育关系,为中药材红花种质资源鉴定和群体遗传学研究奠定基础。利用华大MGISEQ-2000PE150测序平台,双末端测序策略对红花不同花色类型全基因组DNA建库测序,用NOVOPlasty组装叶绿体基因组,采用最大似然法(maximum Likelihood,ML)构建系统进化树。红花3种花色变异类型叶绿体基因组全长均为153 114 bp,完全一致,GC值均为37.8%,具1个典型的四分区域结构,包括1个大单拷贝区(large single copy region,LSC)、1对反向重复区(inverted repeats,IR)和1个短单拷贝区(small single copy,SSC),4个区序列长度分别为84 128、25 194、18 598 bp。红花的叶绿体基因组均有130个基因,其中编码蛋白基因、rRNA基因与tRNA基因的数量均为84、8、38个。系统进化分析表明,红花3种花色变异类型聚在一支,与菜蓟族的铺散矢车菊构成一单系分支,具有100%支持率。叶绿体基因组数据支持红花3种花色变异类型来源为同一种,药用植物红花(所在红花属)隶属于菊科菜蓟族的矢车菊亚族。
红花;叶绿体基因组;组装;系统发育;近缘类群
药用植物红花L.,隶属于菊科(Asteraceae)红花属L.,为一年生草本[1]。红花原产中亚地区,引种后在我国广为栽培,以河南和新疆栽培面积大,所产红花质量好。《中国药典》2020年版规定红花有效成分为羟基红花黄色素A[2]。近年来,化学成分研究表明红花主要含黄酮类物质、脂肪酸、色素、酚酸、挥发油等多种活性成分[3],具有抗炎、抗肿瘤、抗心血管疾病等药理作用,临床应用十分广泛[4]。药用植物红花在栽培过程中,常出现花色分化现象,除常见的橙红色花,还有部分浅黄色和白色花。这些花色变异的类型是种内变异还是来源于同属不同物种,需要进一步的形态和分子验证。目前,药用植物红花的叶绿体基因组虽然已经报道[5],但是对于红花不同花色变异类型的叶绿体基因组组装、基因组特征和系统进化分析等未见报道。
叶绿体是植物最重要的细胞器之一,具有一整套用于光合作用、能量代谢、蛋白质合成及氮、硫同化相关的基因,分布在大小为120~180 kb的环状基因组上,具有结构保守,母性遗传等特点[6-7]。叶绿体基因组一般为闭环双链DNA结构,陆生植物的叶绿体基因组结构通常由1个大单拷贝区域(large single copy,LSC)、1个短单拷贝区域(small single copy,SSC)和2个反向重复区域(inverted repeat,IR)组成[8]。叶绿体基因组拥有相对独立的基因组和遗传序列,并且不像核基因组一般有着复杂的重复序列,其基因序列保守,间隔区变异位点丰富,适宜的进化速率能够为植物不同等级的亲缘关系[9],系统进化关系及遗传多样性研究提供较为可靠的信息[10]。
近年来,叶绿体全基因组测序研究的快速发展为药用植物的分子鉴定研究提供了新的平台及思路[11]。随着测序技术的不断改进,测序平台的不断升级,一系列组装和注释软件如plasmid SPAdes[12]、NOVOPlasty[13]、GetOrganelle[14]等的开发与更新,多种重要的药用植物叶绿体基因组已完成测序和分析,如人参C. A. Meyer[15]、厚朴(Rehder & E. H. Wilson) N. H. Xia & C. Y. Wu[16]、红豆杉(Pilger) Florin[17]、肉苁蓉Ma[18]、三七(Burkill) F. H. Chen ex C. Chow & W. G. Huang[19]、野菊Linnaeus[20]、石斛Lindl.[21]、地黄(Gaert.) Libosch. ex Fisch. et Mey[22]、射干(L.) Redouté[23]等的叶绿体全基因组序列的分析已有相关报道。本研究以红花不同花色变异类型为材料,利用高通量测序方法测定其基因组DNA序列,并对该植物的叶绿体基因组进行组装和注释。分析药用植物红花3种花色类型叶绿体基因组序列特征,IR边界特征,间隔区信息位点的特征,并对红花及其近缘物种共18种(21个样品)植物的叶绿体基因组序列进行系统发育分析,验证其在科级系统发育中的位置,为药用植物红花的种质资源的鉴定、开发和利用提供一定的理论依据。
1 材料
红花L. 3种颜色(橙红色、浅黄色、白色)的新鲜叶片釆集于河南省新乡市原阳县小吴庄村河南农业大学科教园区(35°6′32″N,113°56′34″E),凭证标本号分别为XZ-2020-30、XZ-2020-29、XZ-2020-28。叶片装入取样袋后带回实验室,用无菌水冲洗数次,晾干后置于−80 ℃冰箱备用。样品由河南农业大学农学院中药材系高致明教授和夏至副教授鉴定为菊科红花属的红花。凭证标本保存于河南农业大学标本馆,红花及其近缘物种叶绿体基因组序列来源于NCBI数据库,实验材料详细信息见表1。
2 方法
2.1 DNA的提取和高通量测序
采用北京天根生化植物DNA提取试剂盒(Tiangen Biotech Co.,中国)提取样品红花新鲜叶片的总DNA,利用1%的琼脂糖凝胶电泳检测DNA完整性。样品送至华大生物科技公司(北京)后,进行NanoDrop2000微量分光光度计(Thermo Scientific,美国)检测总DNA的纯度和浓度。MGISEQ-2000 PE150测序平台测序,测序完成后,利用华大自主开发的过滤软件SOAPnuke过滤参数,过滤步骤:(1)过滤接头:测序read匹配上adapter序列的25%或者以上则删除整条read;(2)过滤低质量数据:如果测序read中质量值低于20的碱基占整条read的30%或者以上则删除整条read;(3)去N:如果测序read中N含量占整条read的1%或者以上,则删除整条read;(4)获得Clean reads。数据以FASTQ格式储存,用于后续的拼接和注释。
2.2 叶绿体基因组的拼接和注释
叶绿体基因组拼接釆用NOVOPlasty[13]程序,插入片段大小设为150 bp。过滤后的reads用Geneious 11.0.3[24]拼接软件组装成重叠群,并对组装中的简并碱基,进行人工修正。利用Geneq-Annotation of Organellar(https://chlorobox.mpimpgolm.mpg.de/ geseq.html),结合NCBI上已报道的红花(GenBank登录号:KM207677)注释结果对红花的3个样品叶绿体全基因组进行基因注释,参数为默认值,最后进行手动调整。tRNA用ANAGORNV1.2.38(https:// chlorobox. mpimp golm.mpg.de/geseq.html)预测。注释完成后,提交到NCBI数据库(https://www.ncbi. nlm.nih.gov/ genbank/),GenBank登录号分别为MZ779040(橙红色,XZ2130),MZ779039(浅黄色,XZ2129),MZ779038(白色,XZ2128)。利用在线工具OGDRAW-DRAW Organelle Genome Maps(https: //chlorobox.mpimp-golm.mpg.de/OGDraw. html)绘制叶绿体结构图。
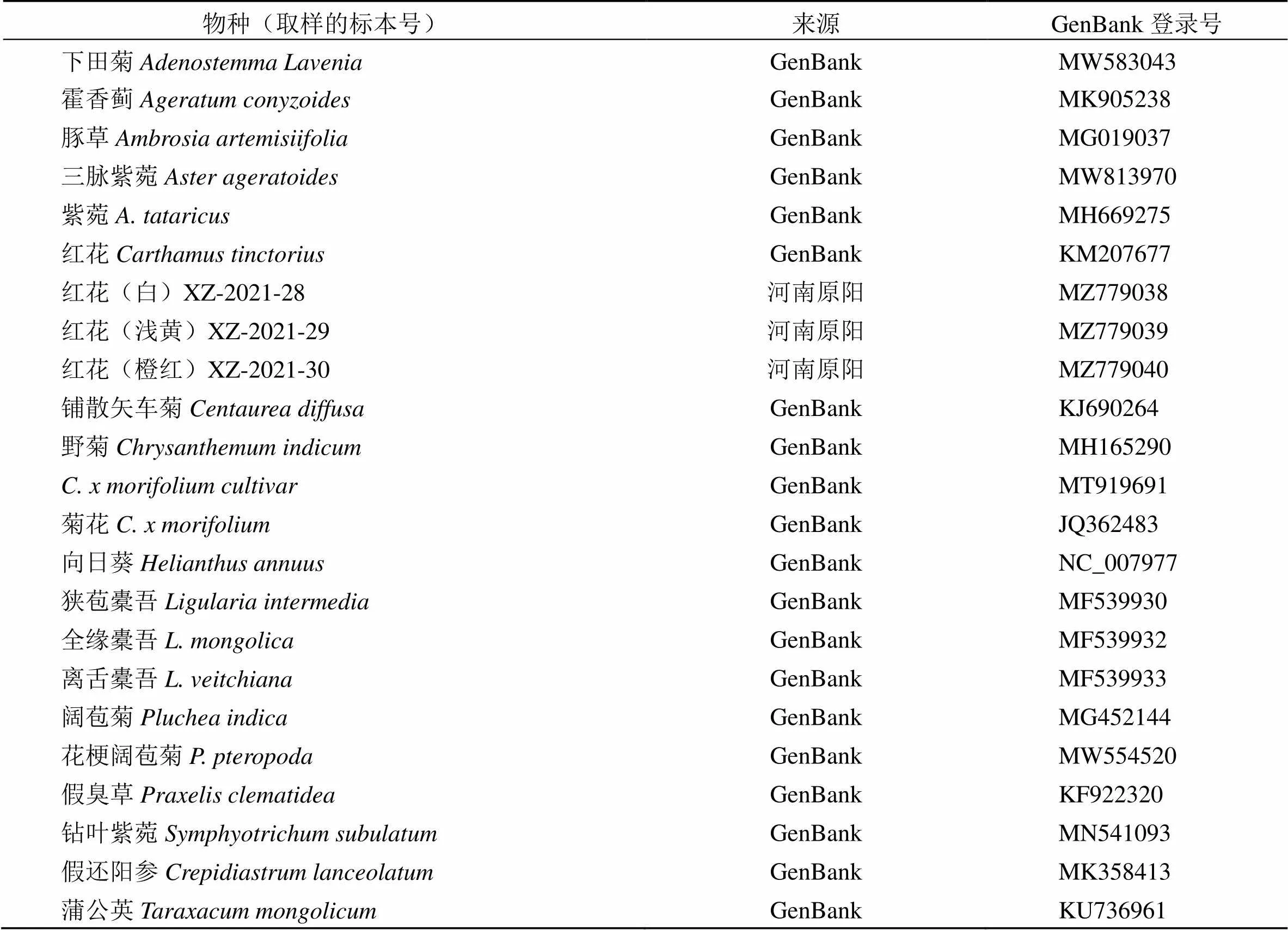
表1 植物样品来源
2.3 叶绿体基因组IR边界的收缩和扩张分析
IR区域在叶绿体基因中具有高度保守性,IR边界的膨胀和收缩被认为是被子植物叶绿体全基因组大小变化的主要机制[25]。在植物进化过程中,IR/SC边界不同程度的扩张和收缩是导致边界和基因组长度多样性的原因[26]。本研究使用Geneious 11.0.3[22]软件获得红花及菊科其他11种植物叶绿体基因组的IRA/IRB、LSC和SSC和边界基因的序列长度,进行比较分析,探讨菊科植物叶绿体基因组IR边界的收缩和扩张特征。并使用Adobe illustrator软件绘制菊科管状花亚科12个属植物叶绿体基因组IR边界对比图。
2.4 叶绿体基因组基因间隔区信息位点分析
相比叶绿体基因编码区,叶绿体基因间隔区在近缘物种间往往具有更高的变异位点,常被用来构建属间、属内、种间物种系统进化发育关系[27]。本研究基于红花及其近缘物种共18种(21个样品)植物的叶绿体基因组序列特征,利用Phylosuite vl.2.1[28]提取红花及其近缘物种共18种(21个样品)植物的叶绿体基因组共有的间隔区。统计这些间隔区的信息位点百分率,为下一步构建菊科属级和属内种间物种系统进化关系提供分子标记。
2.5 系统发育分析
基于菊科(Asteraceae)管状花亚科(Carduoideae)红花及其近缘物种共18种(21个样品)叶绿体基因组数据(表1),同时以菊科(Asteraceae)舌状花亚科(Cichorioideae)假还阳参和蒲公英为外类群,利用phylosuite vl.2.1[26]软件基于MAFFT[29]进行多重比对。系统发育分析采用最大似然法(maximum Likelihood,ML),利用CIPRES Science Gateway服务器(http://www. phylo.org/)中RaxML-HPC2 8.2.12软件[30],采用GTR+GAMMA+I建树模型,构建系统发育树。利用Bootstrap(BS)(1000次重复)检验各分支的支持率。
3 结果与分析
3.1 红花3种花色类型的叶绿体基因组结构特征
橙红色、浅黄色、白色这3种花色的红花,花期一致,植株形态无明显差异,随花期的变化,其颜色一直保持稳定,并且花色差异明显(图1)。测序结果去除接头和低质量的数据,组装和注释后得到橙红色、浅黄色、白色红花的叶绿体基因组。结果表明,红花3种花色类型的叶绿体全基因组均为共价闭合的双链环状分子(图2),全长均为153 114 bp,是一个典型的4分区域结构,包括1个大单拷贝区(LSC)、1对反向重复区(IR)和1个短单拷贝区(SSC),红花3种花色类型的4个区长度均相等,序列长度分别为84 128、25 194、18 598 bp。3种花色的红花在LSC区域、SSC区域和IRs区中GC含量也完全一致,分别为36.0%、31.4%、43.2%。
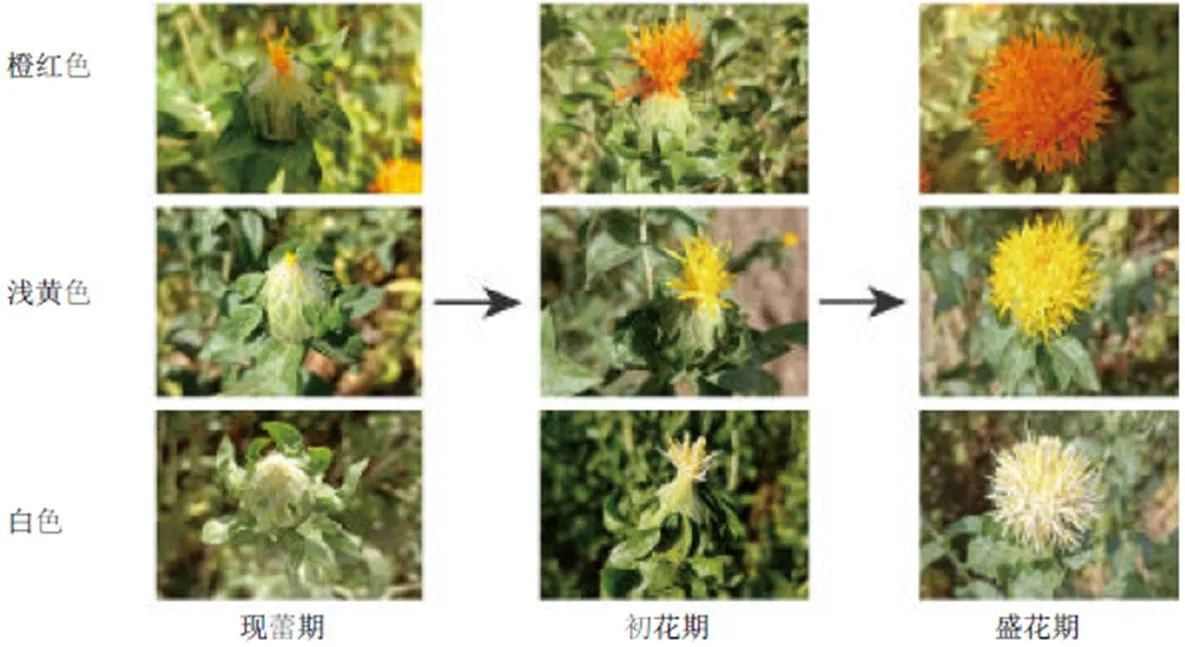
图1 红花(3种花色)不同开花时期的花瓣
3.2 红花3种花色类型叶绿体基因组的组成和特点分析
红花3种花色类型的叶绿体基因组完全一致,均包括130个基因,非重复基因115个,其中84个编码蛋白基因(PCG)、8个rRNA基因与38个tRNA基因。其中,蛋白质编码基因中与自我复制相关基因除rRNA基因和tRNA基因外,还包括13个核糖体小亚基基因、11个核糖体大亚基基因和4个RNA聚合酶亚基基因;光合作用相关的基因有44个,包括12个NADH脱氢酶基因、5个光合系统I基因、14个光合系统II基因、6个细胞色素复合物编码基因、6个ATP合酶基因、1个二磷酸核酮糖羧化酶大亚基基因;此外还有5个其他功能基因及7个未知功能基因(表2)。在tRNA中trnA-UGC、trnI-CAU、trnI-GAU、trnL-、trnN-GUU、trnR-ACG和trnV-GAC各有2个拷贝,trnF-GAA有3个拷贝;4个核糖体RNA(rrn5 rRNA、rrn4.5 rRNA、rrn23 rRNA、rrn16 rRNA)均有2个拷贝,分别位于反向重复区IRA和IRB。核糖体蛋白大小亚基编码的基因中,和这2个基因均有2个拷贝,其余为1个拷贝。NADH脱氢酶亚基中的基因及未知功能蛋白基因和的拷贝数均为2。红花叶绿体基因组中有21个基因有内含子。其中,trnA-UGC、trnI-CAU、trnI-GAU、trnK-UUU、trnL-UAA、trnV-UAC、rps16、rpl2、rpl16、rpl23、rpoC1、ndhA、ndhB、petB、petD和atpF各有1个内含子,而rps12、clpP、ycf3具2个内含子。matK基因位于tmK-UUU基因内,整个编码区为trnK-UUU内含子的一部分,存在序列共用现象。

LSC和SSC:大单拷贝区域、小单拷贝区域;IRA和IRB:2个反向重复区域;内圈深色部分:GC含量

表2 红花叶绿体基因组编码的基因
a和b分别表示含有1个和2个内含子 c-含有2个拷贝基因 d-含有3个拷贝基因
a and b represent 1 and 2 introns respectively, c-contains 2 copy genes, d contains 3 copy genes
3.3 菊科部分物种叶绿体全基因组特征比较分析
菊科植物叶绿体基因组特征的比较分析见表3,红花及其近缘物种共18种(21个样品)植物叶绿体基因组的序列长度范围为150 063~153 318 bp,其中下田菊的叶绿体全基因组长度最短(150 063 bp),钻叶紫菀的叶绿体基因组最长(153 318 bp)。不同花色的红花叶绿体基因组全长完全相同,均为153 114 bp,介于菊科18个物种的叶绿体基因组长度范围之内。红花及其17种近缘植物叶绿体基因组GC含量的范围为37.0%~37.8%,钻叶紫菀的GC含量最低(37.0%),不同花色的红花GC值完全相同,且含量最高(37.8%)。
利用Geneious 11.0.3软件获取红花及17种近缘菊科植物的IR区、LSC区和SSC区序列。结果表明,IR区的序列长度范围为23 776~25 238 bp,其中假臭草的IR区最短(23 776 bp),铺散矢车菊的IR区最长(25 238 bp),不同花色的红花IR区的长度完全一致均为25 194 bp,介于菊科21个样品IR区长度范围之内。LSC区序列长度范围为82 017~94 697 bp,其中下田菊的LSC区长度最短(82 017 bp),紫菀的LSC区长度最长(94 697 bp),不同花色的红花LSC区的长度完全一致均为84 128 bp,介于菊科18个物种LSC区长度范围之内。SSC区序列长度范围介于17 949~18 598 bp,其中豚草的SSC区最短(17 949 bp),不同花色的红花SSC区的长度完全一致且最长(18 598 bp)。
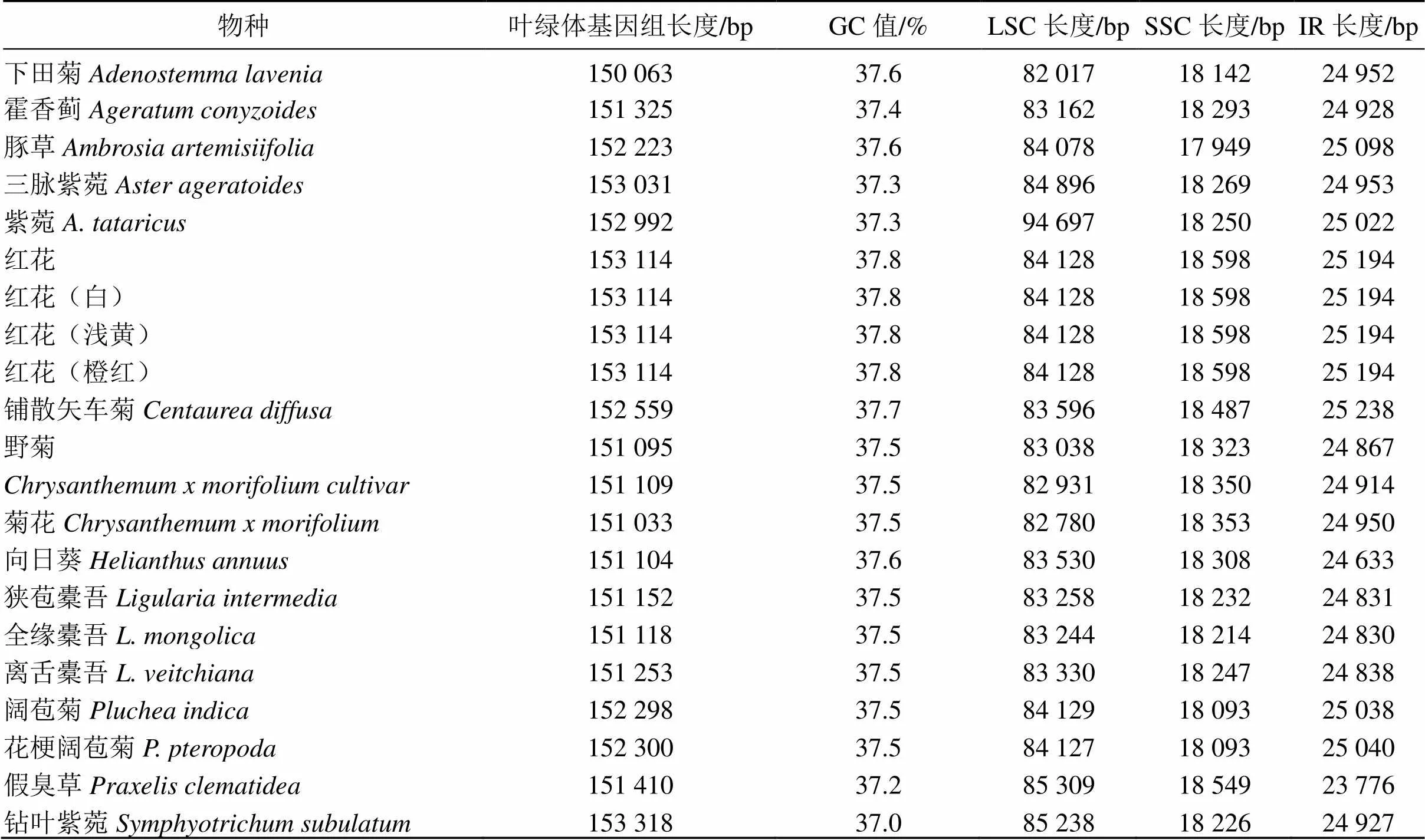
表3 菊科植物18种(21个样品)的叶绿体基因组特征
3.4 红花及菊科11种植物的叶绿体全基因组边界的特征
红花及本研究选取的管状花亚科11个属共12种植物叶绿体基因组的IR-LSC和IR-SSC边界比较显示(图3),不同花色的红花,其边界具有高度的保守性,各边界侧翼基因完全相同,扩张程度也完全一致。红花与其余11种菊科植物叶绿体全基因组相比,边界处具有相同的基因,但是基因的长度仍有差异,部分边界基因也存在变化。LSC/IRb边界(JLB)边界扩张范围显示,红花与其余11种菊科植物相比,JLB边界均位于rps19基因内,但扩张程度稍有差异,红花rps19基因向LSC区域扩张220 bp,向IRb扩张59 bp。SSC/IRb(JSB)边界扩张范围显示,红花与11种菊科植物JSB边界侧翼基因为ycf1和ndhF基因。铺散矢车菊和狭苞橐吾JSB边界位于ycf1基因左翼,距离分别为0、1 bp,而红花与其余9种菊科植物的JSB边界均位于ycf1基因内,但扩张程度稍有差异。SSC/IRa(JSA)边界扩张范围显示,而红花与紫菀、铺散矢车菊、狭苞橐吾和假臭草这5种菊科植物的JSA边界均位于基因内,但扩张程度稍有差异,而下田菊与向日葵JSA边界位于基因左翼、藿香蓟、豚草、菊花、阔苞菊和钻叶紫菀的JSA边界位于基因的右翼;LSC/IRa(JLA)边界扩张范围显示,JLA边界,边界处具有相同的基因,左翼为基因,右翼为基因,但扩张程度稍有差异。此外,铺散矢车菊的叶绿体基因组在IRb区域,显示具有基因,IRa区域存在2基因序列,但是没有注释,因此LSC/IRa边界(JLA)中,基因名称没有显示。

图3 红花及菊科11种植物的叶绿体全基因组边界图
3.5 菊科叶绿体基因组基因间隔区信息位点分析
基于红花及其近缘物种共18种(21个样品)的叶绿体基因组间隔区信息序列特征统计表明(表4),在21个叶绿体基因间隔区中,变异位点百分率变化范围为5.8%~28.8%,最高的trnH-GUG-psbA基因间隔区,其变异位点百分率为28.8%。变异位点百分率超过15%的有6个,分别为ndhC-trnV-UAC、psbE-petL、psbM-trnD-GUC、rps18-rpl20、trnH- GUG-psbA、trnS-GCU-trnC-GCA。变异位点百分率介于10%~15%的有11个,分别为atpB-rbcL、atpH-atpF、atpI-atpH、cemA-petA、psaI-ycf4、psaJ-rpl33、psbK-psbI、rpoC2-rps2、rps18-rpl20、trnC-GCA-petN、trnT-GGU-psbD、ycf4-cemA。变异位点百分率低于10%的有4个,分别为psaA-ycf3、psaA-ycf3、rps2-atpI、trnP-UGG-psaJ。这些变异位点百分率较高的叶绿体基因间隔区,能提供足够多的信息位点,为菊科属间和种间物种进化关系及分子鉴定提供较高的分辨率。

表4 菊科植物18种21个样品叶绿体基因间隔区矩阵位点信息
3.6 菊科叶绿体全基因组系统发育分析结果
基于红花及其近缘物种共18种(21个样品)的叶绿体全基因组的系统发育树结果显示(图4),菊科的管状花亚科类群构成一单系分支,具有100%支持率。同一族取样的物种聚在一起,单系性得到100%支持。在系统树上,取样类群可以分为3个主要分支,A分支是菜蓟族构成的单系分支,具有100%支持率。红花3种不同花色类型与NCBI数据库的红花样品(登录号:KM207677)聚在一支,具有100%支持率,进一步与铺散矢车菊聚在一支,支持率均为100%。分支B包括2个族,紫菀族和千里光族均构成单系分支具有100%的支持率。分支C包括4个族,泽兰族、向日葵族、旋覆花族和看黄菊族构成单系分支具有100%的支持率。
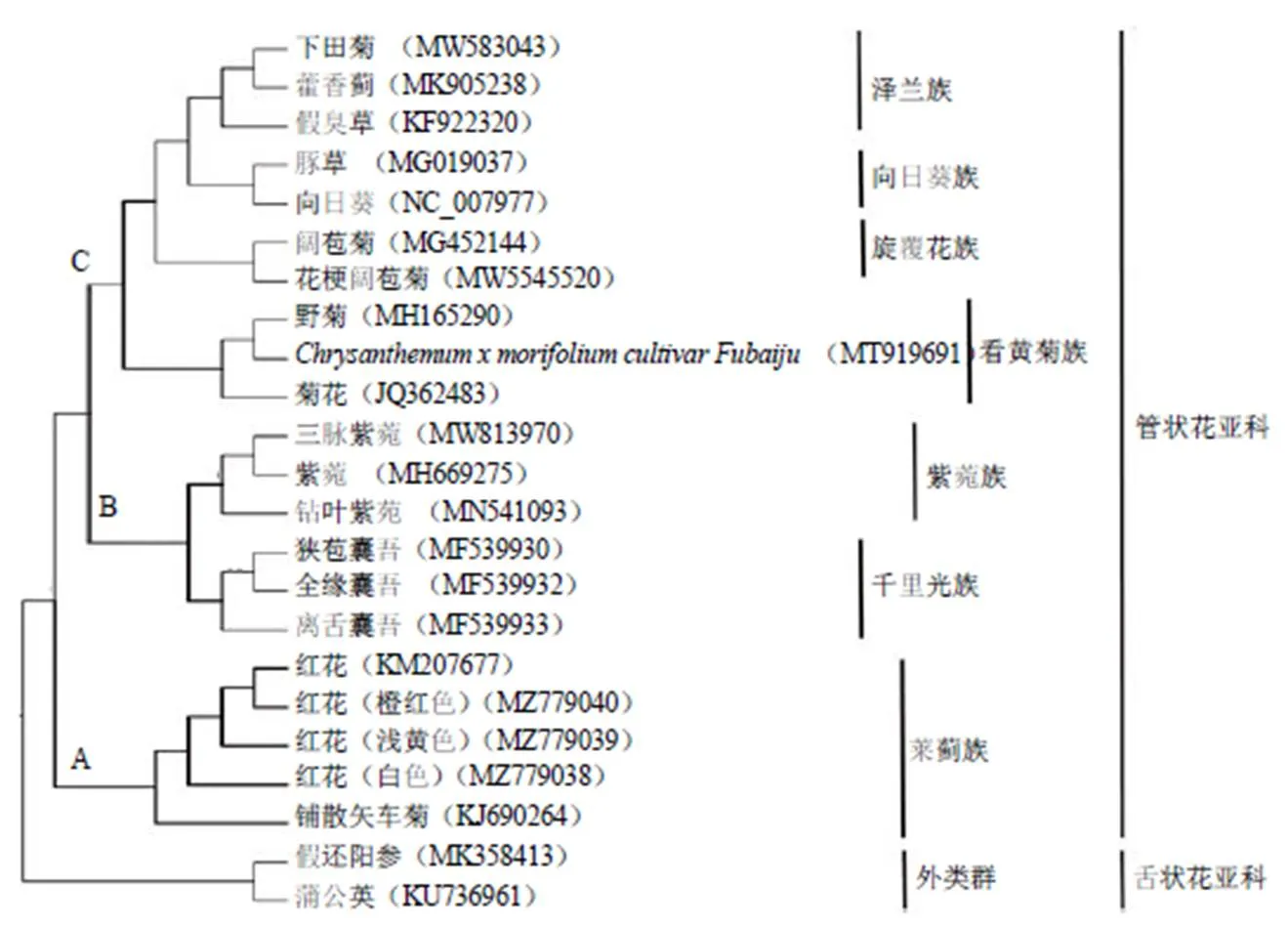
分支上部数值表示ML分析的BS分析对该分支的支持强度(>50%)
4 讨论
红花作为我国传统的大宗药材之一,具有重要的药用和经济价值。本研究完成了药用植物红花3种花色类型的叶绿体基因组的测序,组装与注释。结果表明,红花3种花色的叶绿体基因组序列完全一致,具有典型的4分区域结构(图2),包括1个LSC区、1对IR区和1个SSC区,长度均为153 114 bp。叶绿体基因组数据支持红花3种花色类型,来源为同一种红花,准确鉴定了红花3种花色变异类型的基原物种来源,确保了红花临床用药的安全。红花花色变异可能与花青素生物合成途径相关转录因子分子调控机制有关[31],浅黄色的红花变异类型可能来源于橙红色和白色的变异类型的自然杂交。
对比红花与菊科其他物种的叶绿体基因组,结果表明红花叶绿体基因组长度、IR区和LSC区长度,均位于菊科其他17个物种叶绿体基因组长度范围之内。但红花叶绿体基因组GC值及SSC区序列长度均高于菊科其他17个物种。红花与菊科其他11个属共12种植物的叶绿体基因组IR边界分析(图3)显示,其LSC/IRb边界(JLB),SSC/IRb边界(JSB),SSC/IRa边界(JSA)和LSC/IRa边界(JLA)的侧翼基因完全相同,各边界基因序列的扩张长度略有差异。铺散矢车菊的叶绿体基因组在IRb区域具有基因,同样IRa区域也具有基因,但没有注释,因此LSC/IRa边界(JLA)中,铺散矢车菊基因名称没有显示(图3)。相对于叶绿体基因组编码区基因具有较高的保守性,叶绿体基因组的基因间隔区往往具有丰富的变异位点,基于菊科管状花亚科7个族18种植物(21个样品)的叶绿体基因组间隔区信息序列特征统计表明(表4),变异位点百分率超过15%的基因间隔区有6个,分别为ndhC-trnV-UAC、psbE-petL、psbM-trnD- GUC、rps18-rpl20、trnH-GUG-psbA、trnS-GCU-trnC- GCA基因间隔区。其中trnH-GUG-psbA基因间隔区变异位点最高为28.8%。这些变异位点百分率较高的叶绿体基因间隔区,能提供足够多的信息位点,为菊科属下大范围的物种鉴定,杂交起源,多倍体物种的形成和系统进化分析提供可靠的分子标记片段。
为进一步界定药用植物红花在菊科的系统位置,探讨红花属与其近缘属的系统发育关系。基于菊科18种(21个样品)叶绿体基因组全长构建系统发育树结果显示,取样的管状花亚科所有类群聚在一单系分支具有100%支持率,红花不同花色类型的样品聚在一起具有100%支持率,红花(所在红花属)与铺散矢车菊(所在矢车菊属)聚在一支,构成姐妹群(分支A)具有100%支持率。中国植物志记载[1],菊科菜蓟族的矢车菊亚族(Centaureinae)主要的特征在于它的瘦果有侧生着生面,而药用植物红花(所在红花属)具有典型的瘦果有侧生着生面的形态特征。本研究叶绿体基因组数据分子结果支持传统分类研究结果[1],再次验证药用植物红花(所在红花属)隶属于菜蓟族的矢车菊亚族。
菊科是被子植物中种类最多的一个科,包含大量的药用植物。同时菊科植物也是分布最广的一个类群,是双子叶植物中进化地位最高的一个科,物种形态变异多样关[1]。本研究首次报道了药用植物红花不同花色变异类型的叶绿体全基因组,综合分析菊科药用植物如红花、菊花、野菊花等的叶绿体基因组序列、结构和特征,筛选出一批高变异的叶绿体基因组的间隔区。这为菊科药用植物的分子鉴定,种质资源保护,系统进化关系及遗传多样性研究奠定基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国科学院中国植物志编辑委员会. 中国植物志(第三十八卷) [M]. 北京: 科学出版社, 1986: 56.
[2] 中国药典[S]. 一部. 2020: 1088.
[3] 杨玉霞, 吴卫, 郑有良. 红花研究进展 [J]. 四川农业大学学报, 2004, 22(4): 365-369.
[4] 陈梦, 赵丕文, 孙艳玲, 等. 红花及其主要成分的药理作用研究进展 [J]. 环球中医药, 2012, 5(7): 556-560.
[5] Wu Z H, Liao R, Dong X,. Complete chloroplast genome sequence ofL. from PacBio sequel platform [J]., 2019, 4(2): 2635-2636.
[6] Twyford A D, Ness R W. Strategies for complete plastid genome sequencing [J]., 2017, 17(5): 858-868.
[7] Yu X Y, Zuo L H, Lu D D,. Comparative analysis of chloroplast genomes of fivespecies: Genome comparative and evolution analysis [J]., 2019, 689: 141-151.
[8] Zhang F, Wang T, Shu X,. Complete chloroplast genomes and comparative analyses of L., L. anhuiensis, and(Amaryllidaceae) [J]., 2020, 21(16): E5729.
[9] 邢少辰, Liu C J. 叶绿体基因组研究进展 [J]. 生物化学与生物物理进展, 2008, 35(1): 21-28.
[10] 张靖雯, 姜在民, 蔡靖. 紫丁香与羽叶丁香叶绿体DNA提取方法研究 [J]. 西北林学院学报, 2018, 33(4): 95-99.
[11] 倪梁红, 赵志礼, 米玛. 药用植物叶绿体基因组研究进展 [J]. 中药材, 2015, 38(9): 1990-1994.
[12] Antipov D, Hartwick N, Shen M,. plasmidSPAdes: Assembling plasmids from whole genome sequencing data [J]., 2016, 32(22): 3380-3387.
[13] Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: Deassembly of organelle genomes from whole genome data [J]., 2017, 45(4): e18.
[14] Jin J J, Yu W B, Yang J B,. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes [J]., 2020, 21(1): 241.
[15] Kim K J, Lee H L. Complete chloroplast genome sequences from Korean ginseng (schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants [J]., 2004, 11(4): 247-261.
[16] 李西文, 胡志刚, 林小涵, 等. 基于454FLX高通量技术的厚朴叶绿体全基因组测序及应用研究 [J]. 药学学报, 2012, 47(1): 124-130.
[17] Zhang Y Z, Ma J, Yang B X,. The complete chloroplast genome sequence ofvar.(Taxaceae): Loss of an inverted repeat region and comparative analysis with related species [J]., 2014, 540(2): 201-209.
[18] Li X, Zhang T C, Qiao Q,. Complete chloroplast genome sequence of holoparasite(Orobanchaceae) reveals gene loss and horizontal gene transfer from its host(Chenopodiaceae) [J]., 2013, 8(3): e58747.
[19] 宋菊, 龙月红, 林丽梅, 等. 五加科植物叶绿体基因组结构与进化分析 [J]. 中草药, 2017, 48(24): 5070-5075.
[20] Ma Y P, Zhao L, Zhang W J,. Origins of cultivars of—Evidence from the chloroplast genome and nucleargene [J]., 2020, 58(6): 925-944.
[21] Biswal D, Konhar R, Debnath M,. Chloroplast genome sequence annotation of(Asparagales: Orchidaceae), an endangered medicinal orchid from northeast India [J]., 2017, 9: ecurrents.tol.cf1709613759c2223eb582c0fa694cc7.
[22] Xia Z, Li C C, Hu S W,. The complete chloroplast genome of Chinese medicine cultivar species of(Orobanchaceae) [J]., 2021, 6(1): 290-292.
[23] Li C C, Hu S W, Ding Y N,. The complete chloroplast genome of Chinese medicinal herb(L.) Redouté (Iridaceae) [J]., 2021, 6(2): 331-332.
[24] Kearse M, Moir R, Wilson A,. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data [J]., 2012, 28(12): 1647-1649.
[25] Wang W B, Yu H, Wang J H,. The complete chloroplast genome sequences of the medicinal plant(Oleaceae) [J]., 2017, 18(11): 2288.
[26] Dong W L, Wang R N, Zhang N Y,. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution [J]., 2018, 19(3): 716.
[27] 李世茂. 基于叶绿体DNA基因间隔序列的菊花与近缘种亲缘关系研究 [D]. 武汉: 华中农业大学, 2010.
[28] Zhang D, Gao F L, Jakovlić I,. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies [J]., 2020, 20(1): 348-355.
[29] Katoh K, Standley D M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability [J]., 2013, 30(4): 772-780.
[30] Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies [J]., 2014, 30(9): 1312-1313.
[31] 李莹, 高振蕊, 张驰, 等. 花青素合成途径中分子调控机制的研究进展 [J]. 生态学杂志, 2015, 34(10): 2937-2942.
Characterization and phylogenetic analysis of complete chloroplast genome of different color medicinal plant
DING Yi-ning, BI Guang-yao, HU Sai-wen, ZHOU Yan, ZHOU Yan, LI He-min, XIA Zhi
College of Agronomy, Henan Agricultural University, Zhengzhou 450046, China
In order to provide foundation formolecular identification and population genetics studies, we sequenced and assembled the complete chloroplast genome of three flower color variants,and constructed the phylogenetic relationship between it and its relatives.Genome DNA library was constructed with the paired-end strategy, and MGISEQ-2000PE150 platform was used to sequence in Beijing Genomics Institute (China). The complete chloroplast genome was assembled using NOVOPlasty software, and sequence analysis was performed based on gene annotation results. Phylogenetic analyses were performed using Maximum-Likelihood (ML) methods.The complete chloroplast genome of three different colorshas same length 153 144 bp with a GC content of 37.8%. The chloroplast genome exhibited a typical quadripartite structure, including a large single copy region (LSC), a pair of inverted repeats (IR), and a small single copy (SSC), and the sequence lengths were 84 128 bp, 25 194 bp, and 18 598 bp. The chloroplast genome harbored 130 genes, including 84 protein-coding genes, 8 rRNA genes, and 38 tRNA genes. Phylogenetic analyses result indicates that the three flower color variantsform one clade, and this clade is sister towith bootstrap 100%.Our result supported the three flower color variants offrom the same species. The medicinal plant(genus) belongs to Centaureinae of Cynareae in Asteraceae.
L.; chloroplast genome; assembly; phylogenetic analysis
R286.12
A
0253 - 2670(2023)01 - 0262 - 10
10.7501/j.issn.0253-2670.2023.01.028
2022-07-06
河南省科技攻关项目(222102110137);河南省高等学校重点科研项目计划(22A360010);国家自然科学基金项目(U1404302)
丁怡宁(1997—),女,河南平顶山人,硕士研究生,研究方向为中药资源的分子鉴定。
通信作者:李贺敏,女,副教授,主要从事中药资源的分类鉴定,E-mail: lihemin2002@henau.edu.cn
夏 至,教授,主要从事中药资源的分子鉴定及分子生药学研究。E-mail: xiazhiemail@126.com
[责任编辑 时圣明]
